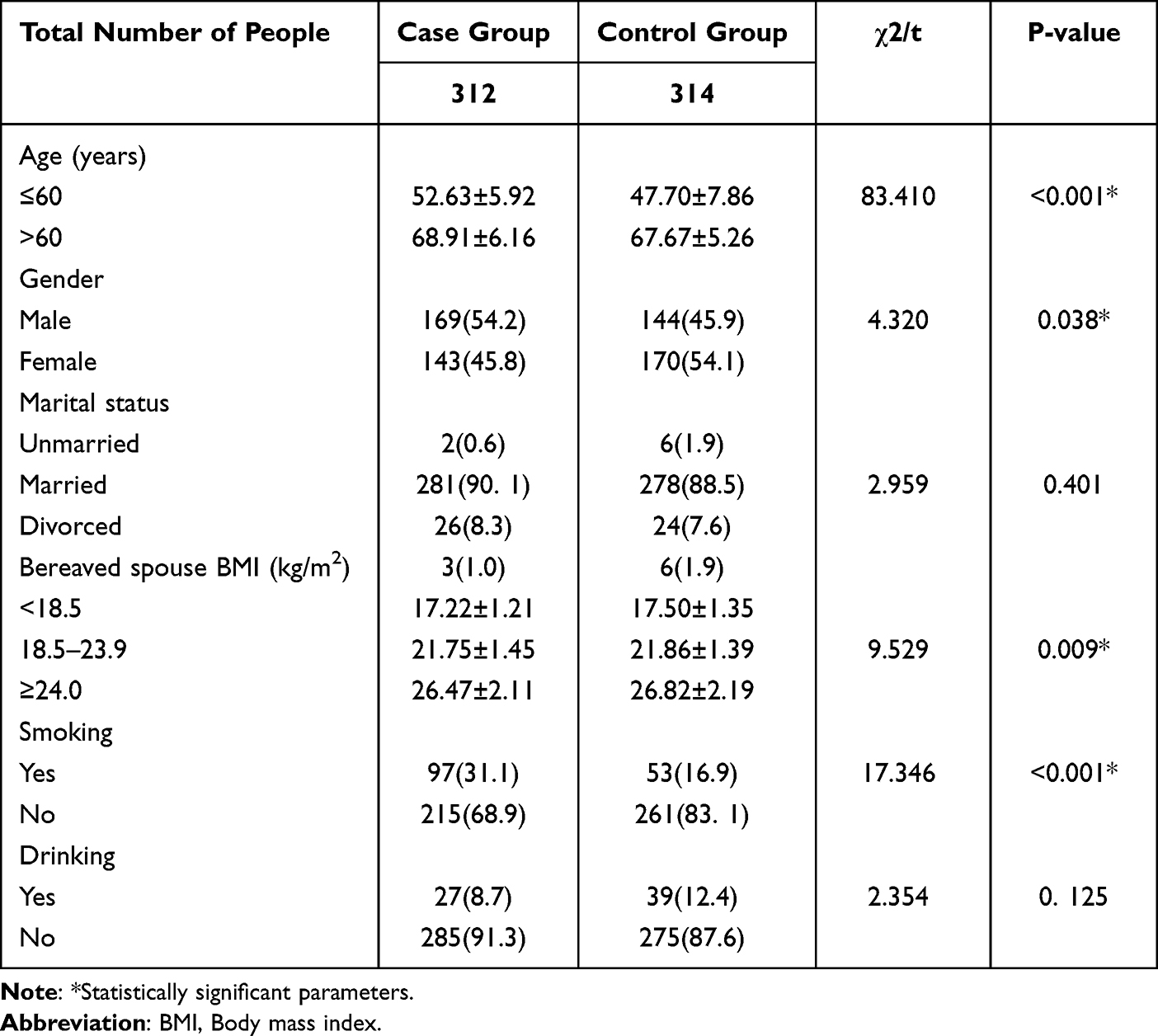
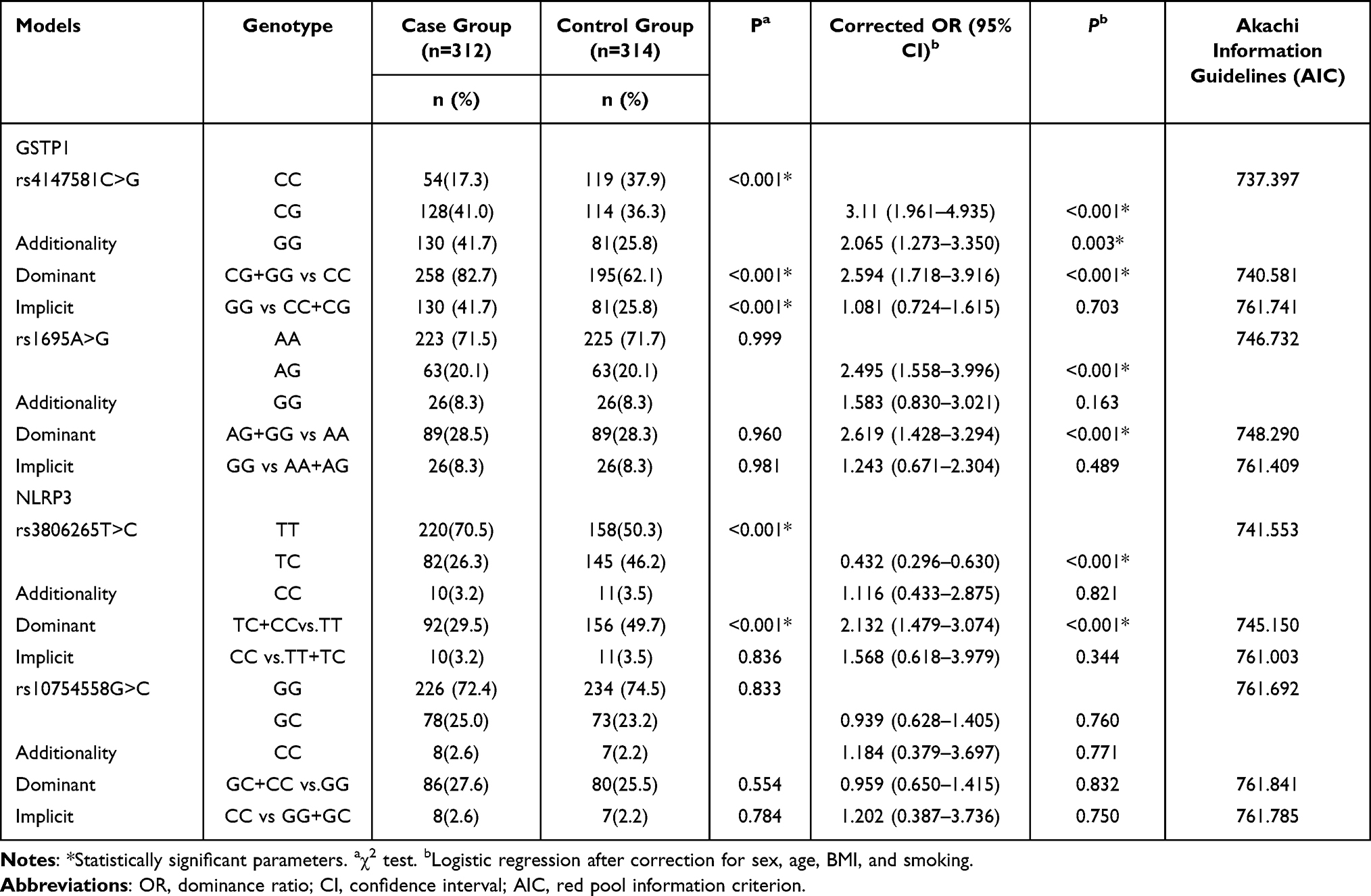
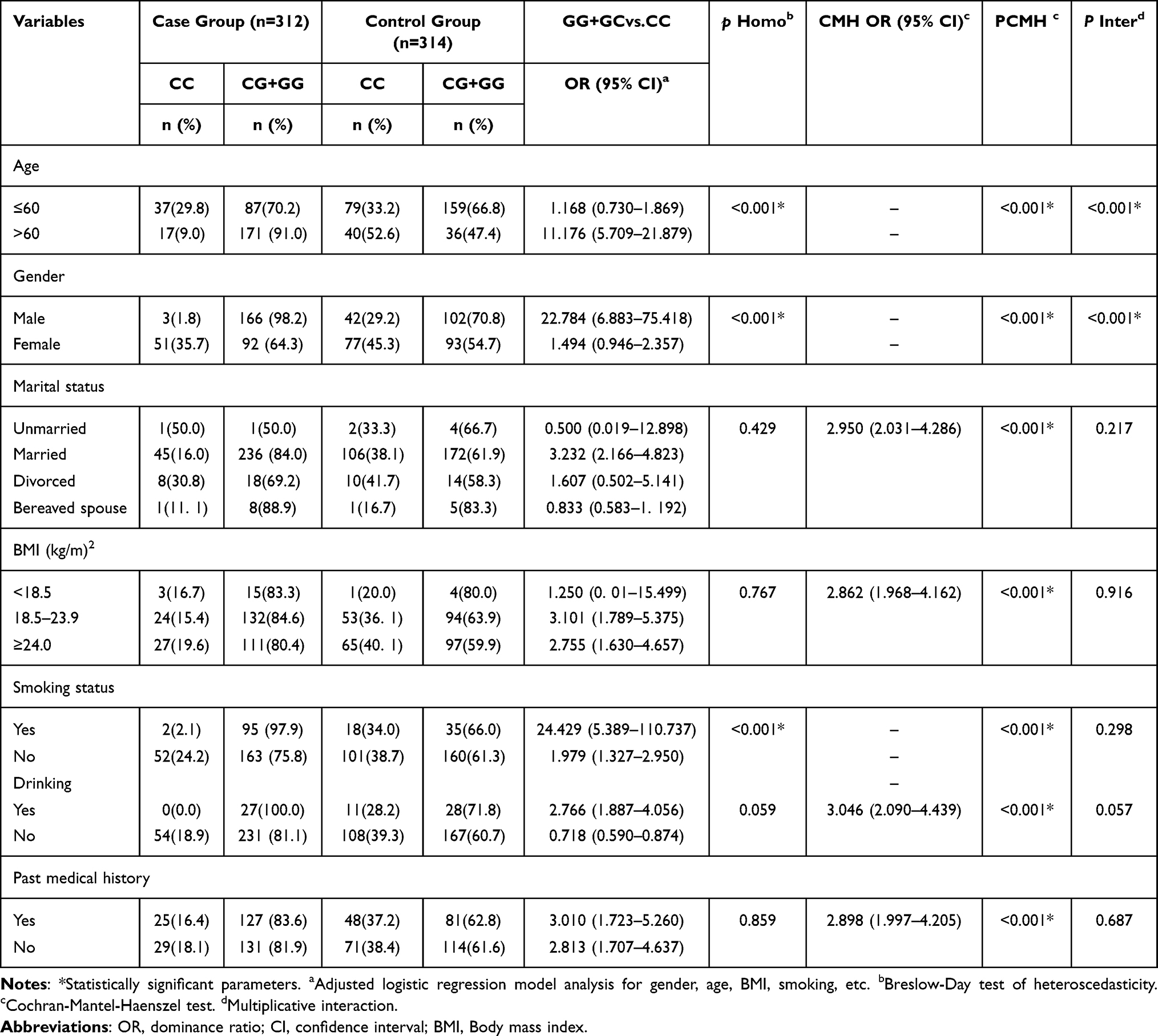
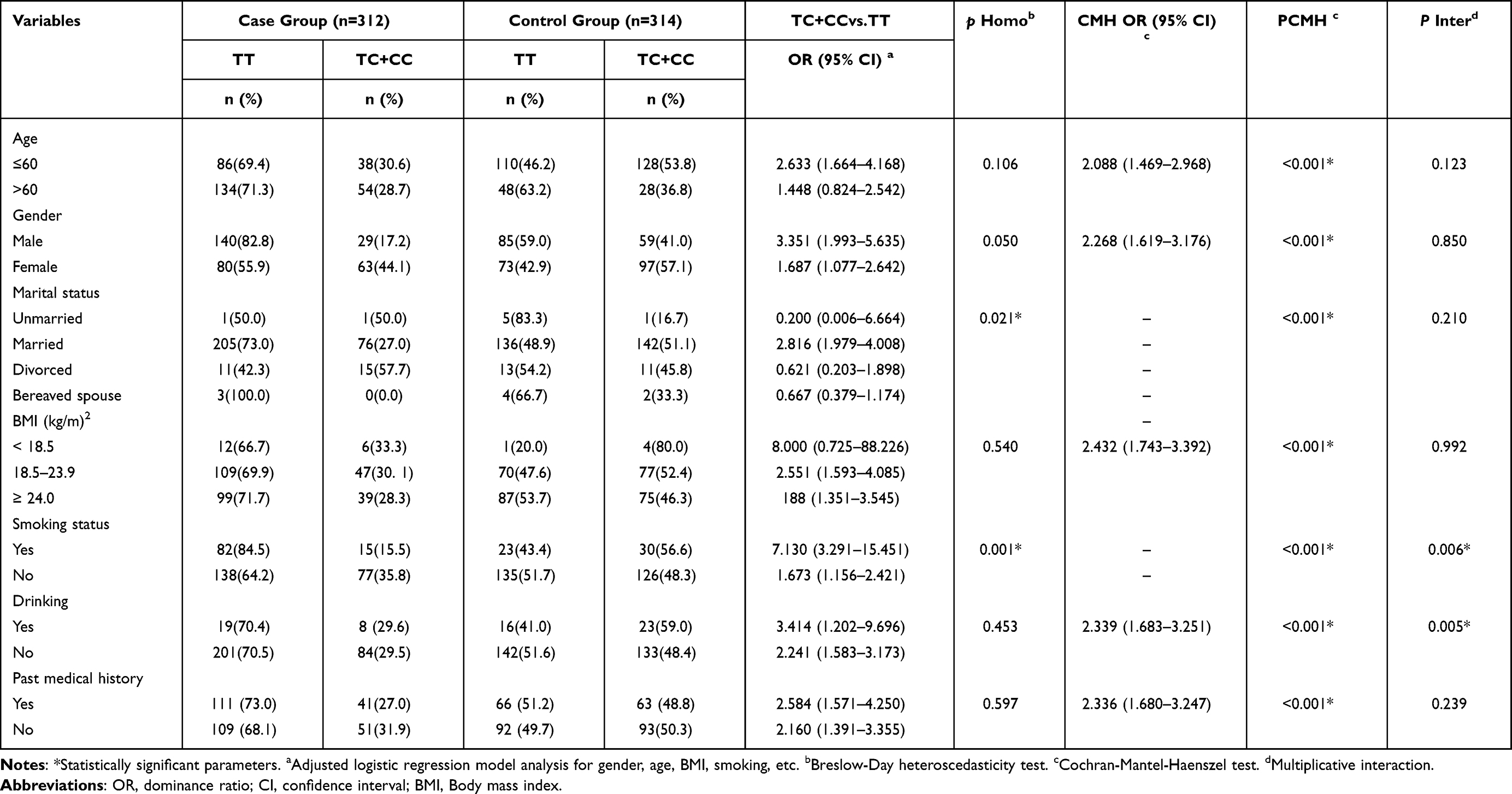
Engineered stone, a popular choice for countertops, has proven
popular due to its aesthetic appeal, cost, durability, and
versatility. However, in recent years there has been focus on the
serious health concerns linked to engineered stone including
long-term respiratory illness and premature death. In this article,
we will delve into what engineered stone is, the serious
respiratory health problems it poses for workers, and the call for
the ban of its use in Australia.
What is engineered stone?
Engineered stone, often known by but not limited to brand names
like Caesarstone, Silestone, or Quantum Quartz, is a popular
material used for kitchen and bathroom countertops, as well as
other interior surfaces. It is made by combining crushed natural
stone, such as quartz, with polymer resins and pigments to create a
durable and attractive surface. The result is a versatile material
with a wide range of colours and patterns that mimics the look of
natural stone at a much cheaper cost, hence the popularity.
What exactly are the health risks linked to engineered
stone?
While engineered stone offers many advantages, there is a
notable downside associated with its production and fabrication.
Engineered stone contains a high concentration of crystalline
silica, a naturally occurring mineral found in quartz, which poses
a significant respiratory health risk when airborne. The fine dust
produced during the cutting, grinding, and polishing of engineered
stone surfaces can be inhaled by workers and lead to severe health
problems, including:
i. Silicosis–
prolonged exposure to respirable crystalline silica dust can lead
to silicosis, an irreversible and often debilitating lung disease.
Silicosis causes scarring of lung tissue, leading to symptoms such
as coughing, breathlessness, and increased susceptibility to
respiratory infections. There is no cure for silicosis and if
developed, life expectancy is diminished.
ii. Lung cancer – inhaling
crystalline silica over an extended period is associated with an
increased risk of lung cancer. Most cases are not curable and
significantly reduce a worker's life expectancy.
iii. Chronic Obstructive Pulmonary Disease
(COPD)– silica exposure can
contribute to the development of COPD, a progressive lung condition
which includes emphysema and chronic bronchitis and is
characterised by breathing difficulties and shortness of
breath.
Silica dust exposure also increases the risk of developing
chronic kidney disease, autoimmune disorders (such as scleroderma
and systemic lupus erythematosus) and other adverse health effects,
including an increased risk of activating latent tuberculosis, eye
irritation and eye damage. The risk posed by engineered stone is
being touted as the new asbestos in terms of the
health ramifications for workers in Australia.
In response to growing concern over the health risks associated
with engineered stone, the NSW government has previously introduced
amendments to the Work Health and Safety Act 2011 (NSW)
which were designed to safeguard the health and well-being of
workers in the engineered stone industry.
These measures included reduced exposure limits, mandatory
health assessments, improved monitoring, and compliance as well as
education and training, and dust control measures which required
employers to implement effective dust control measures, including
proper ventilation, wet cutting methods, and the use of suitable
personal protective equipment.
To date however, persons conducting a business in this industry,
workers and regulators have failed to ensure the health and safety
of all workers working with engineered stone. In particular, the
lack of effective monitoring and compliance, despite some smaller
and sporadic wins, remains a big issue within the industry.
SafeWork Australia (SWA) has called for a
complete ban of the use of engineered stone in Australia. It has
undertaken significant work since 2018 to improve WHS arrangements
to prevent dust diseases including silicosis. This has included
amendments to NSW WHS legislation, however in February 2023 WHS
ministers agreed to SWA's recommendations to address workplace
exposure to respirable crystalline silica through national
awareness and change in behaviour initiatives, and further
regulation for all materials across all industries (which includes
engineered stone).
SWA undertook extensive analysis and consultation on the impacts
of a prohibition on the use of engineered stone and provided its
decision in a report to WHS Ministers on 16 August 2023 for
their consideration. The expert analysis undertaken shows that dust
from engineered stone poses unique hazards, and there is no
evidence that lower silica engineered stone is safer to work with,
meaning there is no safe level of exposure for workers. SWA has
recommended a prohibition on the use of all engineered stone,
irrespective of the crystalline silica content. There is also a
recommendation of the introduction of a licensing scheme to ensure
appropriate controls are in place to protect worker health when
engineered stone already in place needs to be removed, repaired, or
modified.
Silicosis and dust diseases pose an unacceptable health risk to
workers in Australia, and it is important to note that there are
significant financial and non-financial costs associated with being
diagnosed with silicosis or a dust disease, including significant
physical and emotional harm, the reduced ability to work, reduced
quality of life and ultimately premature death of workers. There
are also significant costs to the public health system and in turn
our economy.
SWA recommends urgent government intervention, due to the
disproportionate number of silicosis cases in engineered stone
workers, the younger age of diagnosis of silicosis and dust related
diseases in engineered stone workers, and the impacts on workers,
their families, and the wider community. The decision to prohibit
the use of some or all engineered stone is a matter for WHS
ministers who will meet later this year. It is clear that while
engineered stone revolutionised interior design, the long-term
health risks for workers involved in its fabrication and
installation outweighs the gain.
The content of this article is intended to provide a general
guide to the subject matter. Specialist advice should be sought
about your specific circumstances.
Anyone who has experienced a red air day knows the metallic taste of air pollution that leaves a sting in your nose and lungs. On red air days when pollution hits unhealthy levels people are advised to stay inside and avoid outdoor activities especially the elderly, children, pregnant women, and those who suffer from respiratory illnesses.
Now, guess what is the leading cause of toxic air in most places?
MIAMI - JULY 11: Exhaust flows out of the tailpipe of a vehicle at , "Mufflers 4 Less", July 11, ... [+] 2007 in Miami, Florida. Florida Governor Charlie Crist plans on adopting California's tough car-pollution standards for reducing greenhouse gases under executive orders he plans to sign Friday in Miami. (Photo by Joe Raedle/Getty Images)
Getty Images
Internal combustion engines fueled by gas or diesel are spewing dirty pollution into our lungs and atmosphere. More than two thirds of Americans rely on personal automobiles for day-to-day travel. And transportation is now the United States’ largest source of the greenhouse gas emissions accelerating climate change, with light-duty vehicles alone responsible for nearly 60% of that sector’s climate pollution.
Tackling our pervasive air pollution problem requires cutting tailpipe pollution from the cars we drive. Fortunately, we have a proven tool to make vehicles cleaner so we can all breathe easier: tailpipe emissions standards.
Last week the U.S. Environmental Protection Agency pulled this tool out of the clean air toolbox. The EPA made history by adopting new multi-pollutant rules for light-duty and smaller medium-duty vehicles limiting tailpipe pollution that poisons the air we breathe and accelerates climate change. These updated standards compel automakers to adopt the latest clean technologies to ensure new vehicles will be cleaner than ever before.
Everyone, everywhere should have the choice to make their next car a clean car. Americans who care about reducing pollution deserve the choice to drive electric.
Polluters Want You to Loathe the EPA’s Limits on Pollution
Air pollution harms 36 percent of the U.S. population—or nearly 120 million people. According to the American Lung Association, more than 1 in 3 Americans live in places with unhealthy levels of air pollution, which affects lung development in children and can cause emphysema, asthma, chronic bronchitis, and other respiratory diseases. People of color and lower-income individuals are disproportionately impacted by air pollution.
Anyone who likes breathing cleaner air should celebrate this moment. But corporations that profit from selling the vehicles that pollutes our air and the petroleum that burns a hole in our wallets view these standards as a threat. They say these standards threaten American freedoms and consumer choice.
But the truth is that these updated pollution limits are long overdue and will benefit all Americans by cleaning the air we breathe and giving consumers the choice to get off the expensive fossil fuel rollercoaster.
Consider the American Fuel & Petrochemical Manufacturers’ launch of a “major seven-figure issue campaign across seven critical states—Pennsylvania, Wisconsin, Michigan, Nevada, Arizona, Ohio and Montana—and the Beltway, all aimed at informing Americans about the Biden administration’s efforts to ban new gas, diesel and flex fuel vehicles from the U.S. market.”
In other words, profit-driven corporate polluters responded to the EPA’s updated tailpipe emissions standards by actively manipulating the American public into thinking the standards are unwarranted. Their public disinformation campaign wants people to think the rules are a ban on gas cars.
That is simply false. By law, the EPA does not and cannot ban technologies or modes of transportation. The EPA’s standards are technology-neutral, performance-based, and informed by science and peer-reviewed research.
The Clean Air Act, signed into law in 1970, authorizes and directs the EPA to establish National Ambient Air Quality Standards to protect public health and welfare, to regulate hazardous air pollutant emissions. The Clean Air Act also directs the EPA to regulate emissions from vehicles and engines, and to adapt the standard over time.
The earliest standards for light-duty vehicles required a 90 percent reduction in emissions from hydrocarbons, nitrogen oxides, and carbon monoxide—which drove the development of new engine and emission control technologies, such as the catalytic converter, and a switch to unleaded fuel.
Before the Clean Air Act was signed, our cities choked on air pollution so thick that breathing New York City’s air was as bad as smoking two packs of cigarettes per day, and Los Angeles suffered through unhealthy levels of air pollution more than 200 days a year.
(Original Caption) 6/13/1979-Los Angeles, CA- A lone spectator views a smog-covered downtown Los ... [+] Angeles 6/13. Sun-scorched southern California, still broiling in sweltering 100-degree mercury level now faces a grimy layer of eye-stinging smog. The hot, desert winds responsible for both the current heat wave and clear skies had diminished by late 6/13 and pollution officials issued gloomy predictions of filthy air quality.
Bettmann Archive
50 years later, our air is far cleaner. But despite decades of progress in reducing harmful emissions, air pollution from motor vehicles continues to harm public health, welfare, and the environment. Gas-powered vehicles will always belch ozone, climate pollution, particulate matter, and other toxic chemicals into our air.
These same corporate polluters who are fighting the EPA’s clean air action today have fought against clean air for decades, all for the same reason – profit.
The EPA’s updated standards help level the playing field for more advanced technologies, like battery electric vehicles, to compete in the market. They signal to the auto industry that now is the time to capitalize on fast-falling EV battery costs to deliver more affordable clean vehicle options for all consumers.
Most importantly, the EPA’s rules fix market failures that have allowed corporate fossil fuel profiteers to dump pollution and rising fuel costs on the American public, contaminating the air we breathe while compromising our health and the stability of the climate.
Better Tailpipe Pollution Standards Mean Better Quality of Life
By setting responsible limits on tailpipe pollution the EPA’s updated standards put the U.S. on a new trajectory for cleaner air, better health, and a stable climate. These rules also mean more affordable clean vehicle models on the road for decades to come, saving consumers money every year over the vehicles’ lifetimes. Today, EV models are cheaper to fill than gas vehicles in every state, putting money back in people’s wallets with every trip they take
Data shows electric vehicles are cheaper to fill up than gas vehicles in every state.
Energy Innovation
The EPA’s final rule adopts more stringent emissions standards for criteria pollutants and greenhouse gases for model years 2027-2032 for light-duty vehicles (passenger vehicles), as well as Class 2b and 3 medium-duty vehicles (classes are based on the gross vehicle weight rating; a Ford F-250 is a class 2b vehicle, whereas a Ford F-350 is a class 3 vehicles).
Reduce harmful air pollutants to the tune of 8,700 tons of particulate matter, 36,000 tons of nitrogen oxides, and 150,000 tons of volatile organic compounds in 2055. These pollutants contribute to smog, soot, and bad air days.
Provide $13 billion in annual health benefits.
Reduce approximately 7.2 billion metric tons in net transportation sector CO2 emissions between 2027 and 2055 (the largest source of greenhouse gas emissions at 29 percent of our overall total).
Provide regulatory incentives for vehicle manufacturers to produce engines that emit fewer harmful pollutants, helping more people choose cleaner cars.
Increase zero-emission battery electric vehicle sales over time, ranging from 26% of all new vehicle sales in 2027 to 56% in 2032
Provide $99 billion in annualized net benefits to society through the year 2055; this includes $46 billion in reduced annual fuel costs and nearly $16 billion in reduced maintenance and repair costs for drivers.
Save consumers an average of $6,000 over the lifetime of a new clean vehicle.
Expand consumer choice for American drivers.
Strong Standards Plus New Incentives Will Clean the Air for Generations to Come
The EPA’s updated standards combined with new clean vehicle incentives in the Inflation Reduction Act and new funding in the Bipartisan Infrastructure Law are poised to transform the way we get around. Tax incentives and new funding for vehicles, infrastructure, manufacturing, and the entire clean vehicle supply chain can propel the U.S. toward a transportation transformation.
As it has done for the past 50 years, the EPA is improving air quality. These updated standards reflect significant investments in clean vehicle technologies that the auto industry is already making, and they support growing consumer demand for clean air and a climate safe future.
In time, the updated standards could leave toxic red air days in the rearview mirror - something that will help us all breathe easier.
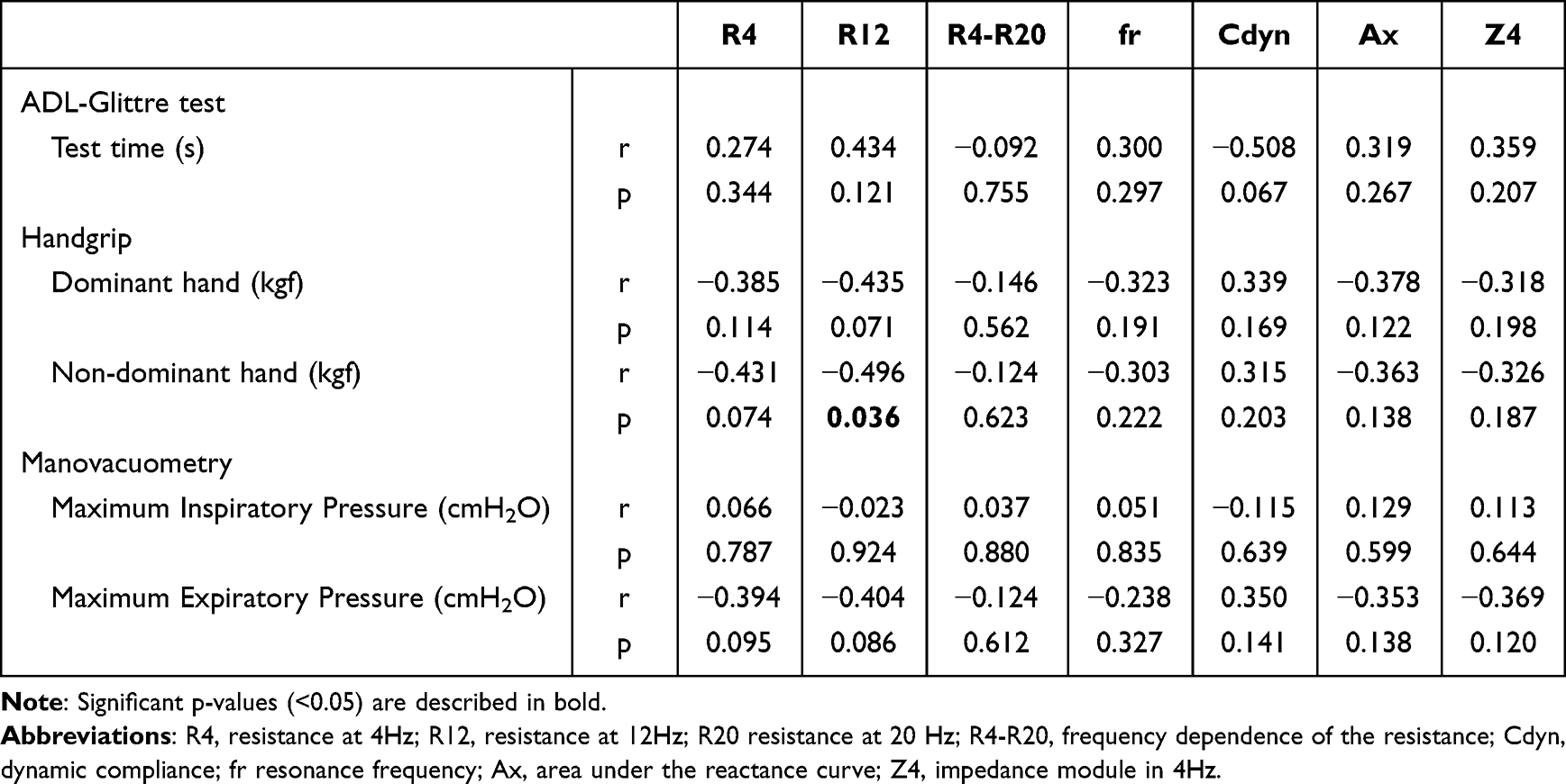
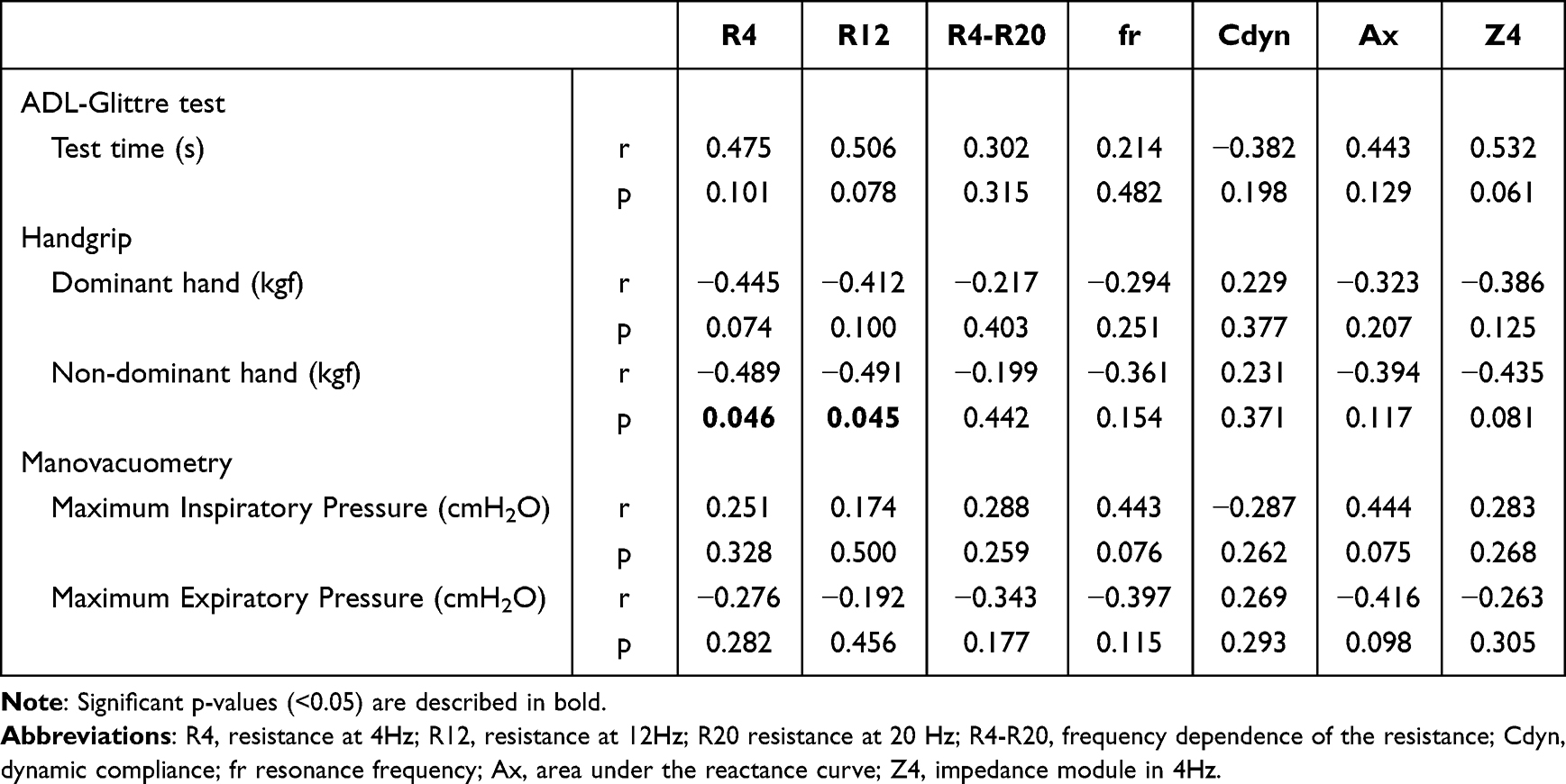
Chronic obstructive pulmonary disease (COPD) is a chronic lung disease with irreversible airflow limitation and a leading cause of death worldwide. COPD is characterized by chronic bronchitis and emphysema, and is associated with malnutrition, muscle weakness, and an increased risk of infection. Although pulmonary tests are considered as the gold standard for COPD diagnosis, they cannot detect early stages of COPD, leading to underdiagnosis. This emphasizes the need for specific biomarkers for early diagnosis, classification, and clinical interventions.
Recent studies suggested that changes in lipids, amino acids, glucose, nucleotides, and microbial metabolites in lungs and intestine, can effectively diagnose early COPD. Metabolomics, a discipline that analyzes different metabolites from body fluids, has emerged as a prominent technique for COPD assessment. However, there are no studies that identify and summarize the metabolites that significantly change during COPD.
A recent review by Dr. Wenqian Wu, Dr. Zhiwei Li, Dr. Tiantian Zhang, and Dr. Hongmei Zhao from the Peking Union Medical College, along with Dr. Yongqiang Wang from 302 Hospital of China Guizhou Aviation Industry Group, and Dr. Chuan Huang at the Chinese Academy of Medical Sciences, provided an in-depth account of the advances in metabolomics of COPD over the last five years, highlighting some potential diagnostic markers and therapeutic targets. Their study was made available online on December 8, 2023, and published in Volume 1, Issue 4 of the journal Chinese Medical Journal Pulmonary and Critical Care Medicine.
Sharing the motivation behind their study, Dr. Tiantian Zhang and Dr. Hongmei Zhao explain, “In addition to altered metabolites from body fluids, increasing evidence has shown that metabolites from pulmonary and intestinal microbes could help us understand the pathogenesis of COPD and the complex regulation underlying this disease.”
Many studies have reported that the three major nutrients, namely protein, lipids, and glucose, along with nucleotide metabolites are closely associated with COPD development and progression.
They found that levels of lipids like sphingolipids and their metabolites, cholesterol, and high-density lipoprotein (HDL), are significantly changed in individuals with COPD. This leads to oxidative stress, inflammation, lipotoxicity, and thus impaired lung function. Various studies suggested that reversing abnormal lipid metabolism and administration of beneficial lipids, might alleviate COPD effects and cardiopulmonary comorbidities.
Dysregulation of amino acid metabolism led to accumulation of harmful metabolites, such as desmosine, isodesmosine, and elastin peptide, which aggravate the damage to the lungs. Furthermore, COPD patients have abnormal levels of amino acids and reduced synthesis capacity of antioxidant carnosine. Some studies suggested that supplementation with amino acids and N-acetylcysteine might be able to regulate amino acid metabolism in COPD.
Glucose metabolism is crucial for energy generation and triggering the immune system. However, this metabolism is dysfunctional in COPD patients resulting in chronic fatigue, muscle weakness and an impaired immune response to pathogens. Further investigation revealed that COPD patients have impaired nucleotide metabolism leading to abnormal levels of adenosine triphosphate (ATP), cyclic adenosine monophosphate (cAMP), and cyclic guanosine monophosphate (cGMP). Nucleotide metabolism impacts on metabolic processes and further studies are warranted to investigate the correlation between nucleotide metabolism and metabolism of lipids, amino acids, and glucose.
Besides metabolic disorders, microorganisms and their metabolites also play a key role in COPD pathogenesis. Individuals with COPD are prone to microbial colonization in their lower respiratory tract. The authors found that both pulmonary and intestinal microbes and their metabolites invade and impact the lungs. Different studies have identified certain common bacteria associated with lung disorders - Streptococcus, Haemophilus influenzae (H. influenzae), Pseudomonas aeruginosa, Campylobacter, to name a few. H. influenzae forms biofilm in the lower airways that acts as a bacterial depot leading to recurrent infections, microbial resistance, evasion of host immune system. Although current studies mainly focus on the bacterial microbiome, fungi and viruses are equally important and demand further studies.
Interestingly chronic lung disorders like COPD impair the gut membrane, increasing gut permeability, microbial movement, and endotoxin release resulting in gut dysbiosis (disease induced imbalance in microbial populations), and a weakened immune response. The intestinal microbiome of COPD patients consists predominantly of microorganisms that reduce lung function, further establishing the correlation between gut microbiome and COPD. On the other hand, gut microorganisms and their metabolites like short chain fatty acids might play a crucial role in alleviating COPD. Thus, gut microflora might be a potential marker for early diagnosis and treatment of COPD.
Overall, this study suggested that efficient regulation of lipid, amino acid, glucose, and nucleotide metabolism along with pulmonary and gut microbial metabolism is essential for COPD management. Dietary modifications to a low-carbohydrate diet and increasing fiber, antioxidant, and vitamin uptake help in COPD prevention.
Dr. Zhang and Dr. Zhao conclude by saying, “Dietary regulation prevents or suppresses respiratory infections by regulating the intestinal microenvironment, which is surprisingly effective in alleviating the symptoms of COPD. We emphasize that intensified dietary management may be among the most feasible methods to improve metabolism in the body.”
***
Reference
Titles of original papers: Advances in metabolomics of chronic obstructive pulmonary disease
Journal: Chinese Medical Journal Pulmonary and Critical Care Medicine
Disclaimer: AAAS and EurekAlert! are not responsible for the accuracy of news releases posted to EurekAlert! by contributing institutions or for the use of any information through the EurekAlert system.
It is learnt that a group of residents from Panaji have reportedly filed two petitions in the Bombay High Court at Goa, urging urgent directions to the government and the Imagine Panaji Smart City Development Corporation (IPSCDL) to alleviate the inconvenience and dust pollution caused by unplanned Smart City projects. The ongoing works has left most of the roads and lanes being dug up. This has caused a lot of dust pollution affecting the health of the residents especially children and the elderly population. Imagine a newborn or an infant breathing in dust-filled air. Dust particles are known for their potential to cause respiratory and cardiovascular health problems. They can also irritate eyes, throat and skin.
Regular dust inhalation can greatly increase one's risk of lung disease and cancer because it weakens the lungs and contributes to disorders like chronic bronchitis. For people with respiratory conditions like asthma, chronic obstructive pulmonary disease (COPD) or emphysema even small increases in dust concentration can make their symptoms worse. According to health experts, air pollution also plays a major part in exacerbating other ailments such as diabetes and sleep apnea. It would hence be in the fitness of things to screen residents of Panaji city for any lung disease due to dust pollution. A white-paper on the respiratory health of residents of Panaji would be welcome.
Adelmo Fernandes, Vasco
Respect sanctity of Basilica
Recently the Central Govt announced a package of Rs 17 crore for the preparation of the works to be undertaken as there will be the Exposition of the Relics of St Francis Xavier fondly known as Goemch Saib.
This happens once in ten years, where the pilgrims and people of other faiths from all over the world and India visit Old Goa to have a lifetime closer look at the Holy Relics of the saint.
The Basilica is under the Archaeological Survey of India and hence the area that has been earmarked as heritage site and has to be maintained by the Govt of India. Catholics should pray that the money sanctioned is used for the purpose that will make the area used as a pilgrimage site and not to promote tourism that will destroy the sanctity of the Spiritual Heritage site. For the Catholics and people of India, it is a blessing and we hope that with the visit of the Pope, Goa and India will be blessed with his prayers giving us hope to live for Jesus and to spread the message of Love and Not hatred. Praise God.
Gregory E D'souza, Siolim
SAG ground finally finds good use
The large open space beside the railway bridge and Taniya Hotel in Vasco, belonging to Sports Authority of Goa (SAG) which was idle for so many years and filled with sewage and tall grass, has finally found some good use for itself. Right after dumping tonnes of mud for the land reclamation process, the ground has generously contributed itself towards various events such as parking space for Vasco Saptah, cricket tournaments, political gatherings, shopping exhibitions etc. For the recent political gathering of the BJP, which took place on that ground itself, it was mentioned by MLA Daji Salkar and the CM that the open space would be converted into a multi-facility sports complex. If this promise finds its manifestation, then this will stand testament to a great leap in the development of the port town, which citizens are eagerly looking forward to.
Milind Jakati, Vasco
Train toll plaza workers to respond to accidents
Despite the availability of modern technology and notwithstanding innumerable first-aid and safety signages, passengers struggle to come to terms with ghastly accidents on highways. The National Highways Authority of India (NHAI) in conjunction with a public sector unit of the ministry of health, is planning to strengthen the Incident Management System (IMS).
The IMS strives to respond to unplanned events and service interruption by restoring the services and events to the originally planned state through a plethora of arms. A few crucial amenities like availability of ambulances will be spruced up so that the " golden hour"— one hour following the accident— is not gone abegging.
Real-time tracking system, too, demands fresh innovation to make it foolproof. As regards to immediate medical aid, the number of trauma centres on highways, if any, require more additions.
It is beyond doubt that all passengers have heard about ‘toll-free phone number’ in exigencies but whether or not the exact number is known to them is anyone's guess. A very vital step undertaken by the NHAI towards ensuring passenger safety is to train the toll plaza workers for instantaneous response to accidents. Education and awareness are two sensitive, and invariable, cogs in the passenger security wheel.
Ganapathi Bhat, Akola
PM’s words just an empty rhetoric
Narendra Modi while on a visit to the North East recently had spoken about protecting our borders which the Congress and earlier governments did not do. Many do not realise that Modi has no problem about protecting borders. The moment our neighbouring country adopts a threatening posture, Modi cedes the territory to them like to China on our northern and north-eastern borders where we gave given lakhs of square kilometers of our land to them. There is no need for protecting borders as far as Modi is concerned!
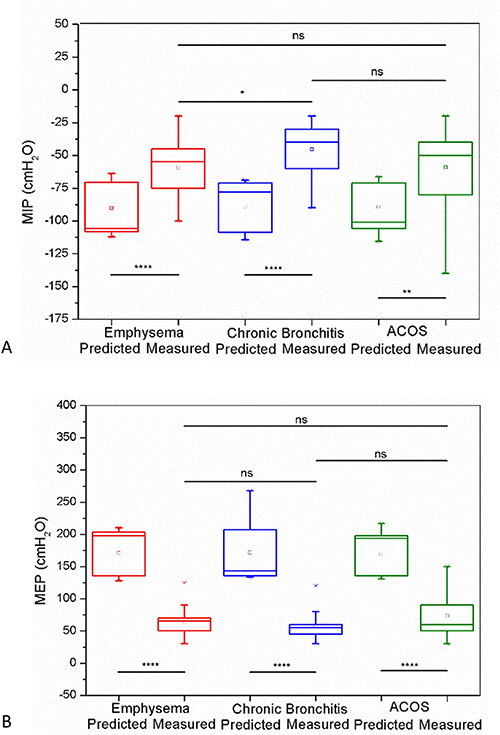
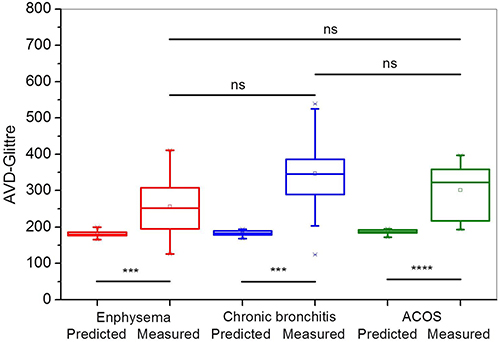
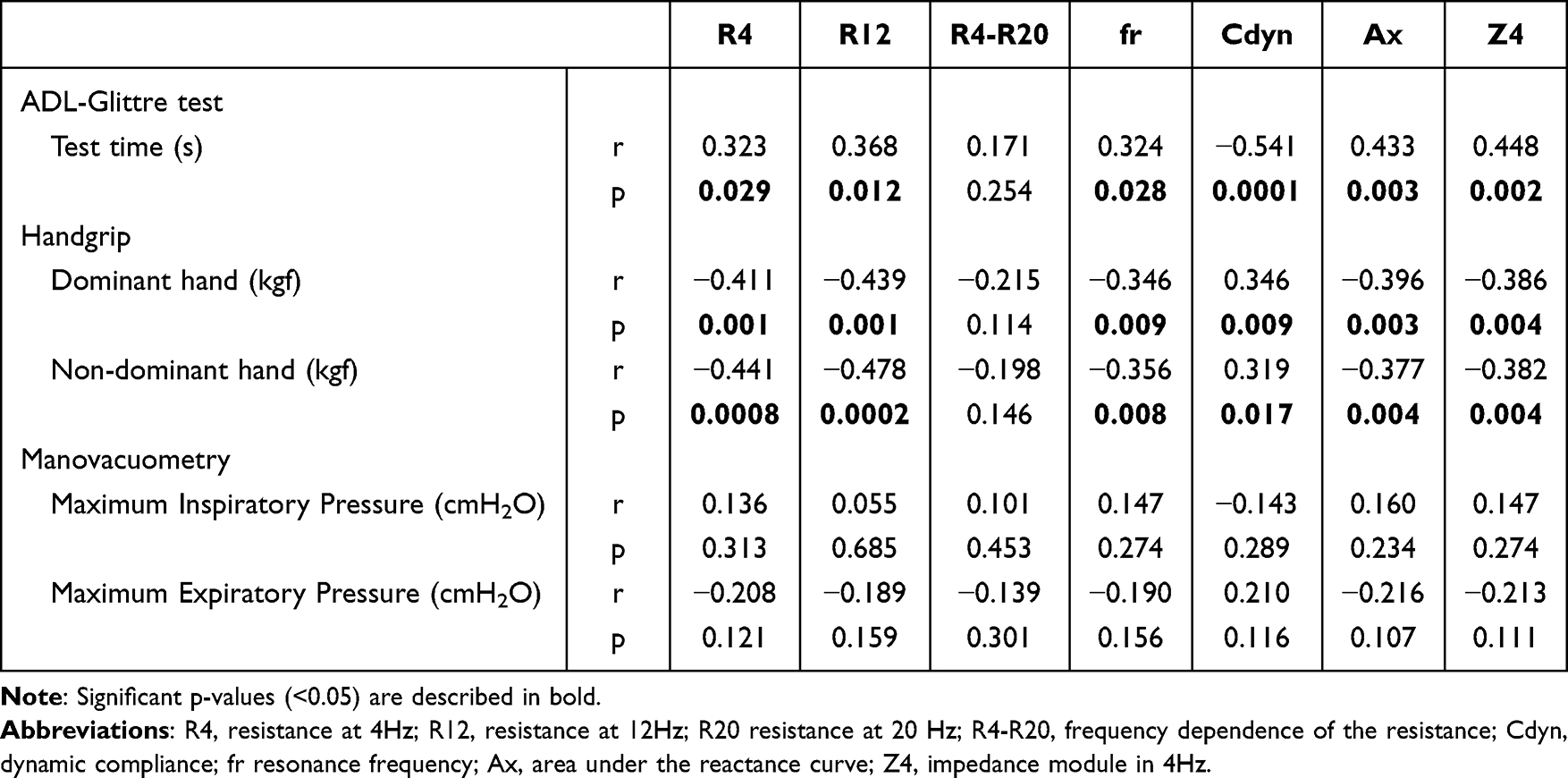
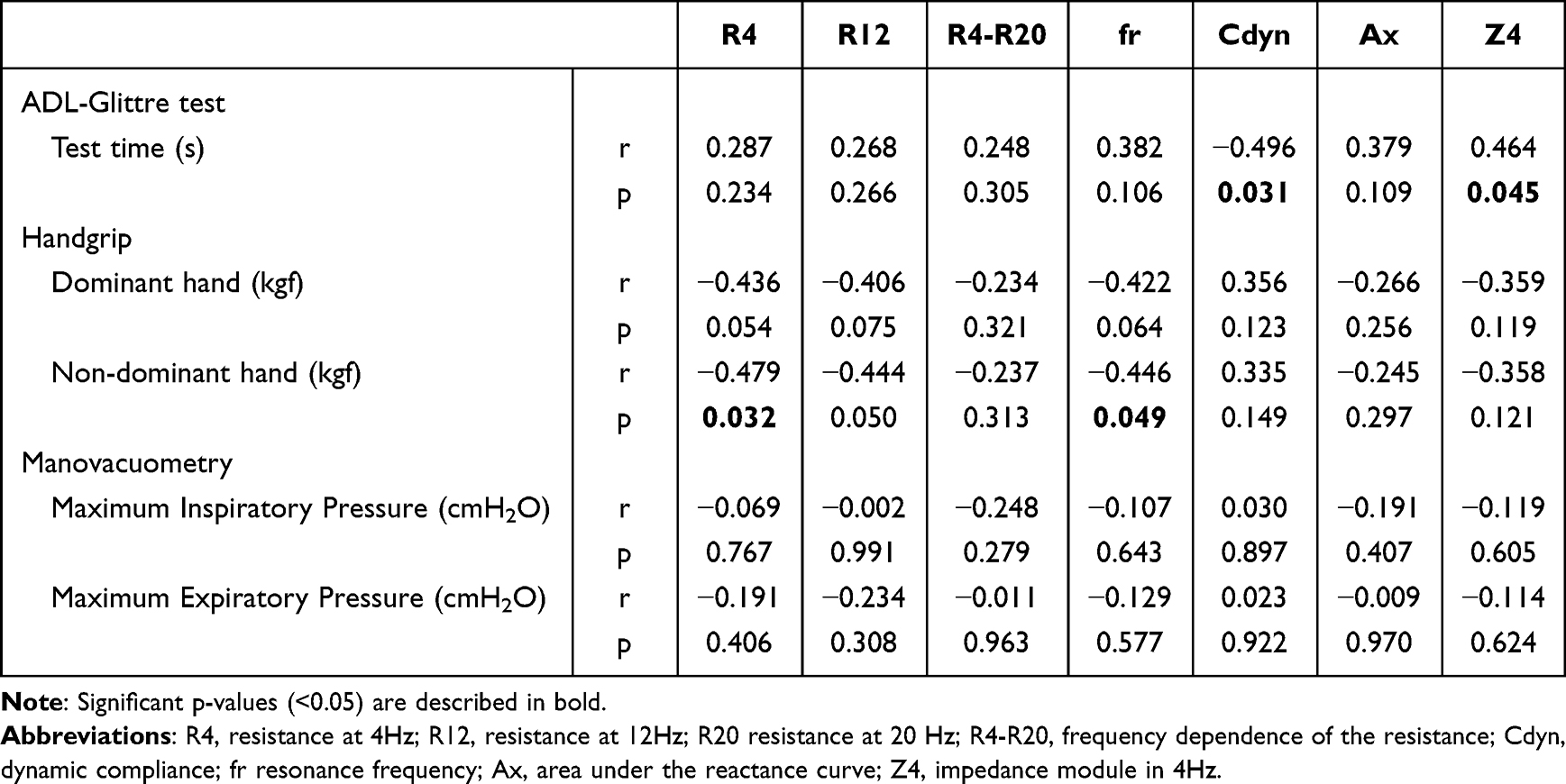
Multiple treatable traits (TTs) were found to be highly prevalent in patients with COPD/advanced emphysema who were eligible for bronchoscopic lung volume reduction (BLVR) using endobronchial valves (EBV), according to study findings published in Respiratory Medicine.
Investigators in The Netherlands conducted a prospective multicenter randomized controlled trial (the SoLVE study; ClinicalTrials.gov Identifier: NCT03474471) to explore the impact of pulmonary rehabilitation on 16 treatable traits in patients with COPD/advanced emphysema receiving EBV treatment. As a secondary outcome, the researchers also characterized TTs associated with severely impaired health-related quality of life (HRQL) using the St. George’s Respiratory Questionnaire (SGRQ).
Eligible patients had a physician diagnosis of COPD/severe emphysema, forced expiratory volume in 1 second (FEV1) of no more than 45% predicted, FEV1/forced vital capacity (FVC) ratio less than 70%, total lung capacity (TLC) greater than 100% predicted, and residual volume greater than 175% predicted. Included patients had a COPD Assessment Test total score of at least 10. Patients with low exercise capacity (6-minute walk test <160 m) and/or severe respiratory failure (partial pressure of carbon dioxide [PaCO2]>8.0kPA and/or partial pressure of oxygen<6.0kPa) were excluded as were those with significant immunodeficiency, bronchiectasis, chronic bronchitis, or previous lobectomy.
The trial included 97 participants (mean [SD] age, 62.4 [6.8] years; 72.9% women). Among the participants, the mean smoking pack years was 39; 58.8% had frequent exacerbations; and 34.0% had at least 1 hospitalization in the previous year.
“
COPD patients with advanced emphysema eligible for BLVR with EBV display a spectrum of treatable traits which were highly prevalent. Having more TTs and more specifically anxiety, depression or fatigue, is associated with a worse HRQL.
TTs assessed were: (1) severe dyspnea; (2) very severe airflow limitation; (3) frequent exacerbations; (4) poor exercise capacity; (5) low physical activity; (6) hypoxemia; (7) hypercapnia; (8) underweight; (9) obesity; (10) low muscle mass; (11) decreased bone mineral density; (12) impaired handgrip force; (13) impaired quadriceps force; (14) severe fatigue; (15) anxiety; and (16) depression.
The mean (SD) TTs per participant was 8.1 (2.5; range 2-15); the most prevalent TTs included low physical activity (95%), poor exercise capacity (94%), and severe fatigue (75%).
Overall, participants were characterized by severe lung hyperinflation, severe airflow limitation, and poor HRQL (median total SGRQ score was 60).
When participants were stratified by low vs high SGRQ total score (less than 60 points vs 60 or greater points), the researchers found that the most significant predictors of having a higher SGRQ total score were severe fatigue, depression, and anxiety. A significant although moderate positive correlation was found between the sum of TTs present in a participant and that participant’s SGRQ total score (r=0.53; P <.001).
Study limitations include the strict inclusion/exclusion criteria, which may have affected the prevalence of specific TTs; lack of examination of some significant TTs (eg, persistent systemic inflammation, adherence to pharmacotherapy, family/social support); and lack of a comparator severe COPD group not eligible for BLVR-EBV.
The study authors concluded that “COPD patients with advanced emphysema eligible for BLVR with EBV display a spectrum of treatable traits which were highly prevalent. Having more TTs and more specifically anxiety, depression or fatigue, is associated with a worse HRQL. Findings of this study advocate a multidimensional assessment and management of this specific COPD phenotype.”
Expanded savings programs build on company’s longstanding commitment to addressing barriers to access and affordability for patients
AstraZeneca announced it will expand the savings programs for its entire US inhaled respiratory portfolio, helping eligible patients pay no more than $35 per month for their medicine.* Expanding the savings programs will help make its inhalers more affordable to the most vulnerable patients living with asthma and chronic obstructive pulmonary disease (COPD), including those who are uninsured and underinsured.
Pascal Soriot, Chief Executive Officer, AstraZeneca, said: “AstraZeneca’s expanded savings programs build on our longstanding commitment to addressing barriers to access and affordability for patients living with respiratory diseases to ultimately help patients lead healthier lives. We remain dedicated to addressing the need for affordability of our medicines, but the system is complex and we cannot do it alone. It is critical that Congress bring together key stakeholders to help reform the healthcare system so patients can afford the medicines they need, not just today, but for the future.”
Starting June 1, 2024, eligible patients will pay no more than $35 per month for all AstraZeneca US inhaled respiratory medicines, including:
AIRSUPRA® (albuterol and budesonide)
BEVESPI AEROSPHERE® (glycopyrrolate and formoterol fumarate) Inhalation Aerosol
BREZTRI AEROSPHERE® (budesonide, glycopyrrolate, and formoterol fumarate) Inhalation Aerosol
SYMBICORT® (budesonide and formoterol fumarate dihydrate) Inhalation Aerosol
In addition, AstraZeneca substantially reduced the list price of SYMBICORTon January 1, 2024. The Company will continue to provide discounts and rebates off the list price to help patients afford its inhaled respiratory medicines.
For more than 50 years, AstraZeneca has served respiratory patients by investing in the research and development of new drug-device combinations, as well as next-generation biologics and novel mechanisms to address the vast unmet needs of these chronic, often debilitating diseases. AstraZeneca remains dedicated to transforming patient outcomes, while ensuring access and affordability of our innovative medicines.
*Terms and conditions apply. Government restrictions exclude people enrolled in federal government insurance programs from co-pay support.
IMPORTANT SAFETY INFORMATION
AIRSUPRA® (albuterol and budesonide)
Contraindications: Hypersensitivity to albuterol, budesonide, or to any of the excipients
Deterioration of Asthma: Asthma may deteriorate acutely over a period of hours or chronically over several days or longer. If the patient continues to experience symptoms after using AIRSUPRA or requires more doses of AIRSUPRA than usual, it may be a marker of destabilization of asthma and requires evaluation of the patient and their treatment regimen
Paradoxical Bronchospasm: AIRSUPRA can produce paradoxical bronchospasm, which may be life threatening. Discontinue AIRSUPRA immediately and institute alternative therapy if paradoxical bronchospasm occurs. It should be recognized that paradoxical bronchospasm, when associated with inhaled formulations, frequently occurs with the first use of a new canister
Cardiovascular Effects: AIRSUPRA, like other drugs containing beta2-adrenergic agonists, can produce clinically significant cardiovascular effects in some patients, as measured by pulse rate, blood pressure, and/or other symptoms. If such effects occur, AIRSUPRA may need to be discontinued. In addition, beta-agonists have been reported to produce electrocardiogram (ECG) changes, such as flattening of the T wave, prolongation of the QTc interval, and ST-segment depression. Therefore, AIRSUPRA, like all sympathomimetic amines, should be used with caution in patients with cardiovascular disorders, especially coronary insufficiency, cardiac arrhythmias, and hypertension
Do Not Exceed Recommended Dose: Clinically significant cardiovascular effects and fatalities have been reported in association with excessive use of inhaled sympathomimetic drugs
Hypersensitivity Reactions, Including Anaphylaxis: Can occur after administration of albuterol sulfate and budesonide, components of AIRSUPRA, as demonstrated by cases of anaphylaxis, angioedema, bronchospasm, oropharyngeal edema, rash, and urticaria. Discontinue AIRSUPRA if such reactions occur
Risk of Sympathomimetic Amines with Certain Coexisting Conditions: AIRSUPRA, like all therapies containing sympathomimetic amines, should be used with caution in patients with convulsive disorders, hyperthyroidism, or diabetes mellitus and in patients who are unusually responsive to sympathomimetic amines
Hypokalemia: Beta-adrenergic agonist medicines may produce significant hypokalemia in some patients. The decrease in serum potassium is usually transient, not requiring supplementation
Immunosuppression and Risk of Infections: Due to possible immunosuppression from the use of inhaled corticosteroids (ICS), potential worsening of infections could occur. Use with caution. A more serious or fatal course of chickenpox or measles can occur in susceptible patients
Oropharyngeal Candidiasis: Has occurred in patients treated with ICS agents. Monitor patients periodically. Advise patients to rinse his/her mouth with water, if available, without swallowing after inhalation
Hypercorticism and Adrenal Suppression: May occur with very high doses in susceptible individuals. If such changes occur, consider appropriate therapy
Reduction in Bone Mineral Density: Decreases in bone mineral density have been observed with long-term administration of ICS. For patients at high risk for decreased bone mineral density, assess initially and periodically thereafter
Glaucoma and Cataracts: Have been reported following the long-term administration of ICS, including budesonide, a component of AIRSUPRA
Effects on Growth: Orally inhaled corticosteroids, including budesonide, may cause a reduction in growth velocity when administered to pediatric patients. The safety and effectiveness of AIRSUPRA have not been established in pediatric patients, and AIRSUPRA is not indicated for use in this population
Most common adverse reactions (incidence ≥ 1%) are headache, oral candidiasis, cough, and dysphonia
Drug Interactions: AIRSUPRA should be administered with caution to patients being treated with:
Strong cytochrome P450 3A4 inhibitors (may cause systemic corticosteroid effects)
Short-acting bronchodilators (concomitant use of additional beta-agonists with AIRSUPRA should be used judiciously to prevent beta-agonist overdose)
Beta-blockers (may block pulmonary effects of beta-agonists and produce severe bronchospasm)
Diuretics or non-potassium-sparing diuretics (may potentiate hypokalemia or ECG changes). Consider monitoring potassium levels
Monoamine oxidase inhibitors (MAOI) or tricyclic antidepressants (Use AIRSUPRA with extreme caution; may potentiate effect of albuterol on the cardiovascular system)
Use AIRSUPRA with caution in patients with hepatic impairment, as budesonide systemic exposure may increase. Monitor patients with hepatic disease
BEVESPI AEROSPHERE® (glycopyrrolate and formoterol fumarate) Inhalation Aerosol
CONTRAINDICATIONS
All long-acting beta2-adrenergic agonists (LABAs), including formoterol fumarate, are contraindicated in patients with asthma without use of an inhaled corticosteroid. BEVESPI is not indicated for the treatment of asthma. BEVESPI is contraindicated in patients with hypersensitivity to glycopyrrolate, formoterol fumarate, or to any component of the product.
WARNINGS AND PRECAUTIONS
The safety and efficacy of BEVESPI AEROSPHERE in patients with asthma have not been established. BEVESPI AEROSPHERE is not indicated for the treatment of asthma
Use of LABAs as monotherapy (without inhaled corticosteroids [ICS]) for asthma is associated with an increased risk of asthma-related death. These findings are considered a class effect of LABA monotherapy. When LABAs are used in fixed-dose combination with ICS, data from large clinical trials do not show a significant increase in the risk of serious asthma-related events (hospitalizations, intubations, death) compared to ICS alone. Available data do not suggest an increased risk of death with use of LABAs in patients with chronic obstructive pulmonary disease (COPD)
BEVESPI should not be initiated in patients with acutely deteriorating COPD, which may be a life-threatening condition
BEVESPI should not be used for the relief of acute symptoms (ie, as rescue therapy for the treatment of acute episodes of bronchospasm). Acute symptoms should be treated with an inhaled short-acting beta2-agonist (SABA)
BEVESPI should not be used more often or at higher doses than recommended, or with other LABAs, as an overdose may result
If paradoxical bronchospasm occurs, discontinue BEVESPI immediately and institute alternative therapy
If immediate hypersensitivity reactions occur, in particular, angioedema, urticaria, or skin rash, discontinue BEVESPI at once and consider alternative treatment
BEVESPI can produce a clinically significant cardiovascular effect in some patients, as measured by increases in pulse rate, blood pressure, or symptoms. If such effects occur, BEVESPI may need to be discontinued
Use with caution in patients with convulsive disorders, thyrotoxicosis, diabetes mellitus, ketoacidosis, and in patients who are unusually responsive to sympathomimetic amines
Be alert to hypokalemia and hyperglycemia
Worsening of narrow-angle glaucoma or urinary retention may occur. Use with caution in patients with narrow-angle glaucoma, prostatic hyperplasia, or bladder-neck obstruction, and instruct patients to contact a physician immediately if symptoms occur
ADVERSE REACTIONS
The most common adverse reactions with BEVESPI (≥2% and more common than placebo) were cough, 4.0% (2.7%) and urinary tract infection, 2.6% (2.3%).
DRUG INTERACTIONS
Use caution if administering additional adrenergic drugs because the sympathetic effects of formoterol may be potentiated
Concomitant treatment with xanthine derivatives, steroids, or diuretics may potentiate any hypokalemic effect of formoterol
Use with caution in patients taking non-potassium-sparing diuretics, as the ECG changes and/or hypokalemia may worsen with concomitant beta2-agonists
The action of adrenergic agonists on the cardiovascular system may be potentiated by monoamine oxidase inhibitors, tricyclic antidepressants, or other drugs known to prolong the QTc interval. Therefore, BEVESPI should be used with extreme caution in patients being treated with these agents
Use beta-blockers with caution as they not only block the therapeutic effects of beta-agonists, but may produce severe bronchospasm in patients with COPD
Avoid co-administration of BEVESPI with other anticholinergic-containing drugs as this may lead to an increase in anticholinergic adverse effects
INDICATION
BEVESPI AEROSPHERE is a combination of glycopyrrolate, an anticholinergic, and formoterol fumarate, a long-acting beta2-adrenergic agonist (LABA), indicated for the maintenance treatment of patients with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema.
LIMITATION OF USE
Not indicated for the relief of acute bronchospasm or for the treatment of asthma.
BREZTRI AEROSPHERE® (budesonide, glycopyrrolate, and formoterol fumarate) Inhalation Aerosol
BREZTRI is contraindicated in patients who have a hypersensitivity to budesonide, glycopyrrolate, formoterol fumarate, or product excipients
BREZTRI is not indicated for treatment of asthma. Long-acting beta2-adrenergic agonist (LABA) monotherapy for asthma is associated with an increased risk of asthma-related death. These findings are considered a class effect of LABA monotherapy. When a LABA is used in fixed-dose combination with ICS, data from large clinical trials do not show a significant increase in the risk of serious asthma-related events (hospitalizations, intubations, death) compared with ICS alone. Available data do not suggest an increased risk of death with use of LABA in patients with COPD
BREZTRI should not be initiated in patients with acutely deteriorating COPD, which may be a life-threatening condition
BREZTRI is NOT a rescue inhaler. Do NOT use to relieve acute symptoms; treat with an inhaled short-acting beta2-agonist
BREZTRI should not be used more often than recommended; at higher doses than recommended; or in combination with LABA-containing medicines, due to risk of overdose. Clinically significant cardiovascular effects and fatalities have been reported in association with excessive use of inhaled sympathomimetic drugs
Oropharyngeal candidiasis has occurred in patients treated with orally inhaled drug products containing budesonide. Advise patients to rinse their mouths with water without swallowing after inhalation
Lower respiratory tract infections, including pneumonia, have been reported following ICS. Physicians should remain vigilant for the possible development of pneumonia in patients with COPD as the clinical features of pneumonia and exacerbations frequently overlap
Due to possible immunosuppression, potential worsening of infections could occur. Use with caution. A more serious or fatal course of chickenpox or measles can occur in susceptible patients
Particular care is needed for patients transferred from systemic corticosteroids to ICS because deaths due to adrenal insufficiency have occurred in patients during and after transfer. Taper patients slowly from systemic corticosteroids if transferring to BREZTRI
Hypercorticism and adrenal suppression may occur with regular or very high dosage in susceptible individuals. If such changes occur, consider appropriate therapy
Caution should be exercised when considering the coadministration of BREZTRI with long-term ketoconazole and other known strong CYP3A4 Inhibitors. Adverse effects related to increased systemic exposure to budesonide may occur
If paradoxical bronchospasm occurs, discontinue BREZTRI immediately and institute alternative therapy
Anaphylaxis and other hypersensitivity reactions (eg, angioedema, urticaria or rash) have been reported. Discontinue and consider alternative therapy
Use caution in patients with cardiovascular disorders, especially coronary insufficiency, as formoterol fumarate can produce a clinically significant cardiovascular effect in some patients as measured by increases in pulse rate, systolic or diastolic blood pressure, and also cardiac arrhythmias, such as supraventricular tachycardia and extrasystoles
Decreases in bone mineral density have been observed with long-term administration of ICS. Assess initially and periodically thereafter in patients at high risk for decreased bone mineral content
Glaucoma and cataracts may occur with long-term use of ICS. Worsening of narrow-angle glaucoma may occur, so use with caution. Consider referral to an ophthalmologist in patients who develop ocular symptoms or use BREZTRI long term. Instruct patients to contact a healthcare provider immediately if symptoms occur
Worsening of urinary retention may occur. Use with caution in patients with prostatic hyperplasia or bladder-neck obstruction. Instruct patients to contact a healthcare provider immediately if symptoms occur
Use caution in patients with convulsive disorders, thyrotoxicosis, diabetes mellitus, and ketoacidosis or unusually responsive to sympathomimetic amines
Be alert to hypokalemia or hyperglycemia
Most common adverse reactions in a 52-week trial (incidence ≥ 2%) were upper respiratory tract infection (5.7%), pneumonia (4.6%), back pain (3.1%), oral candidiasis (3.0%), influenza (2.9%), muscle spasms (2.8%), urinary tract infection (2.7%), cough (2.7%), sinusitis (2.6%), and diarrhea (2.1%). In a 24-week trial, adverse reactions (incidence ≥ 2%) were dysphonia (3.3%) and muscle spasms (3.3%)
BREZTRI should be administered with extreme caution to patients being treated with monoamine oxidase inhibitors and tricyclic antidepressants, as these may potentiate the effect of formoterol fumarate on the cardiovascular system
BREZTRI should be administered with caution to patients being treated with:
Strong cytochrome P450 3A4 inhibitors (may cause systemic corticosteroid effects)
Adrenergic drugs (may potentiate effects of formoterol fumarate)
Beta-blockers (may block bronchodilatory effects of beta-agonists and produce severe bronchospasm)
Anticholinergic-containing drugs (may interact additively). Avoid use with BREZTRI
Use BREZTRI with caution in patients with hepatic impairment, as budesonide and formoterol fumarate systemic exposure may increase. Patients with severe hepatic disease should be closely monitored
INDICATION
BREZTRI AEROSPHERE is indicated for the maintenance treatment of patients with chronic obstructive pulmonary disease (COPD).
LIMITATIONS OF USE
Not indicated for the relief of acute bronchospasm or for the treatment of asthma.
SYMBICORT® (budesonide and formoterol fumarate dihydrate) Inhalation Aerosol
Use of long-acting beta2-adrenergic agonists (LABA) as monotherapy (without inhaled corticosteroids [ICS]) for asthma is associated with an increased risk of asthma-related death. Available data from controlled clinical trials also suggest that use of LABA as monotherapy increases the risk of asthma-related hospitalization in pediatric and adolescent patients. These findings are considered a class effect of LABA. When LABA are used in fixed dose combination with ICS, data from large clinical trials do not show a significant increase in the risk of serious asthma-related events (hospitalizations, intubations, death) compared to ICS alone
SYMBICORT is NOT a rescue medication and does NOT replace fast-acting inhalers to treat acute symptoms
SYMBICORT should not be initiated in patients during rapidly deteriorating episodes of asthma or COPD
Patients who are receiving SYMBICORT should not use additional formoterol or other LABA for any reason
Localized infections of the mouth and pharynx with Candida albicans has occurred in patients treated with SYMBICORT. Patients should rinse the mouth after inhalation of SYMBICORT
Lower respiratory tract infections, including pneumonia, have been reported following the administration of ICS
Due to possible immunosuppression, potential worsening of infections could occur. A more serious or even fatal course of chickenpox or measles can occur in susceptible patients
It is possible that systemic corticosteroid effects such as hypercorticism and adrenal suppression may occur, particularly at higher doses. Particular care is needed for patients who are transferred from systemically active corticosteroids to ICS. Deaths due to adrenal insufficiency have occurred in asthmatic patients during and after transfer from systemic corticosteroids to less systemically available ICS
Caution should be exercised when considering administration of SYMBICORT in patients on long-term ketoconazole and other known potent CYP3A4 inhibitors
As with other inhaled medications, paradoxical bronchospasm may occur with SYMBICORT
Immediate hypersensitivity reactions may occur, as demonstrated by cases of urticaria, angioedema, rash, and bronchospasm
Excessive beta-adrenergic stimulation has been associated with central nervous system and cardiovascular effects. SYMBICORT should be used with caution in patients with cardiovascular disorders, especially coronary insufficiency, cardiac arrhythmias, and hypertension
Long-term use of ICS may result in a decrease in bone mineral density (BMD). Since patients with COPD often have multiple risk factors for reduced BMD, assessment of BMD is recommended prior to initiating SYMBICORT and periodically thereafter
ICS may result in a reduction in growth velocity when administered to pediatric patients
Glaucoma, increased intraocular pressure, and cataracts have been reported following the administration of ICS, including budesonide, a component of SYMBICORT. Close monitoring is warranted in patients with a change in vision or history of increased intraocular pressure, glaucoma, or cataracts
In rare cases, patients on ICS may present with systemic eosinophilic conditions
SYMBICORT should be used with caution in patients with convulsive disorders, thyrotoxicosis, diabetes mellitus, ketoacidosis, and in patients who are unusually responsive to sympathomimetic amines
Beta-adrenergic agonist medications may produce hypokalemia and hyperglycemia in some patients
The most common adverse reactions ≥3% reported in asthma clinical trials included nasopharyngitis, headache, upper respiratory tract infection, pharyngolaryngeal pain, sinusitis, pharyngitis, rhinitis, influenza, back pain, nasal congestion, stomach discomfort, vomiting, and oral candidiasis
The most common adverse reactions ≥3% reported in COPD clinical trials included nasopharyngitis, oral candidiasis, bronchitis, sinusitis, and upper respiratory tract infection
SYMBICORT should be administered with caution to patients being treated with MAO inhibitors or tricyclic antidepressants, or within 2 weeks of discontinuation of such agents
Beta-blockers may not only block the pulmonary effect of beta-agonists, such as formoterol, but may produce severe bronchospasm in patients with asthma
ECG changes and/or hypokalemia associated with nonpotassium-sparing diuretics may worsen with concomitant beta-agonists. Use caution with the coadministration of SYMBICORT
INDICATIONS
SYMBICORT is indicated for the treatment of asthma in patients 6 years and older not adequately controlled on a long-term asthma-control medication such as an ICS or whose disease warrants initiation of treatment with both an ICS and LABA (also see DOSAGE AND ADMINISTRATION).
SYMBICORT 160/4.5 is indicated for the maintenance treatment of airflow obstruction in patients with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema, and to reduce COPD exacerbations.
SYMBICORT is NOT indicated for the relief of acute bronchospasm.
Asthma is a chronic, inflammatory respiratory disease with variable symptoms that affects as many as 262 million people worldwide,1 including approximately 25 million in the US.2
Patients with asthma experience recurrent breathlessness and wheezing, which varies over time, and in severity and frequency.3 These patients are at risk of severe exacerbations regardless of their disease severity, adherence to treatment or level of control.4-5
There are an estimated 136 million asthma exacerbations globally per year,6 including approximately 10 million in the US2; these are physically threatening and emotionally significant for many patients7 and can be fatal.3,8
Inflammation is central to both asthma symptoms4 and exacerbations.9 Many patients experiencing asthma symptoms use a SABA (e.g., albuterol) as a rescue medicine10-12; however, taking a SABA alone does not address inflammation, leaving patients at risk of severe exacerbations,13 which can result in impaired quality of life,14 hospitalization15 and frequent oral corticosteroid (OCS) use.15 Treatment of exacerbations with as few as 1-3 short courses of OCS are associated with an increased risk of adverse health conditions including type 2 diabetes, depression/anxiety, renal impairment, cataracts, cardiovascular disease, pneumonia and fracture.16 International recommendations from the GINA no longer recommend SABA alone as the preferred rescue therapy.3
About COPD
COPD refers to a group of lung diseases, including chronic bronchitis and emphysema, that cause airflow blockage and breathing-related problems.17 Affecting an estimated 16 million Americans, COPD is the third leading cause of death due to chronic disease and the sixth overall leading cause of death in the US.18-19
About AIRSUPRA®
AIRSUPRA (albuterol and budesonide), formerly known as PT027, is a first-in-class SABA/ICS rescue treatment for asthma in the US, to be taken as needed. It is an inhaled, fixed-dose combination rescue medication containing albuterol (also known as salbutamol), a SABA, and budesonide, a corticosteroid, and has been developed in a pMDI using AstraZeneca’s Aerospheredelivery technology.
The FDA approval of AIRSUPRA was based on MANDALA and DENALI Phase III trials (Approval press release). In MANDALA, AIRSUPRA significantly reduced the risk of severe exacerbations compared to albuterol in patients with moderate-to-severe asthma when used as an as-needed rescue medication in response to symptoms. For patients treated with AIRSUPRA 180 mcg/160 mcg the annualized total systemic corticosteroids dose when compared with albuterol 180 mcg was statistically significantly different, with a reduction in mean annualized dose of 40 mg per patient. In DENALI, AIRSUPRA significantly improved lung function compared to the individual components albuterol and budesonide in patients with mild to moderate asthma.
About BEVESPI AEROSPHERE®
BEVESPI AEROSPHERE (glycopyrronium and formoterol fumarate) is a fixed-dose dual bronchodilator in a pMDI, combining glycopyrronium, a long-acting muscarinic antagonist (LAMA), and formoterol fumarate, a long-acting beta2-agonist (LABA). PMDIs are an important choice for COPD patients where limited lung function, advanced age and reduced dexterity or cognition are significant considerations for patients to achieve therapeutic benefits from their medicines. BEVESPI AEROSPHERE is the only LABA/LAMA with Aerosphere delivery technology. Results from an imaging trial have shown that BEVESPI AEROSPHERE effectively delivers medicine to both the large and small airways.
About BREZTRI AEROSPHERE®
BREZTRI AEROSPHERE (budesonide, glycopyrrolate, and formoterol fumarate) is a single-inhaler, fixed-dose triple-combination of formoterol fumarate, a LABA, glycopyrronium bromide, a LAMA, with budesonide, an ICS, and delivered in a pressurized metered-dose inhaler. BREZTRI AEROSPHEREis approved to treat COPD in more than 50 countries worldwide including the US, EU, China and Japan, and is currently being studied in Phase III trials for asthma.
About SYMBICORT®
Symbicort (budesonide and formoterol fumarate dihydrate) is the number one ICS/LABA combination therapy in asthma and chronic obstructive pulmonary disease (COPD) in China. It is a combination formulation containing budesonide, an ICS that treats underlying inflammation, and formoterol, a LABA with a fast onset of action, in a single inhaler. Symbicort was launched in 2000 and is approved in approximately 120 countries to treat asthma and/or COPD either as Symbicort Turbuhaler or Symbicort pMDI (pressurised metered-dose inhaler).
About AstraZeneca in Respiratory & Immunology
Respiratory & Immunology, part of BioPharmaceuticals, is one of AstraZeneca’s main disease areas and is a key growth driver for the Company.
AstraZeneca is an established leader in respiratory care with a 50-year heritage. The Company aims to transform the treatment of asthma and COPD by focusing on earlier biology-led treatment, eliminating preventable asthma attacks, and removing COPD as a top-three leading cause of death. The Company’s early respiratory research is focused on emerging science involving immune mechanisms, lung damage and abnormal cell-repair processes in disease and neuronal dysfunction.
With common pathways and underlying disease drivers across respiratory and immunology, AstraZeneca is following the science from chronic lung diseases to immunology-driven disease areas. The Company’s growing presence in immunology is focused on five mid- to late-stage franchises with multi-disease potential, in areas including rheumatology (including systemic lupus erythematosus), dermatology, gastroenterology, and systemic eosinophilic-driven diseases. AstraZeneca’s ambition in Respiratory & Immunology is to achieve disease modification and durable remission for millions of patients worldwide.
AstraZeneca
AstraZeneca is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialization of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit www.astrazeneca-us.com and follow us on social media @AstraZeneca.
About AZ&Me™
AstraZeneca’s patient assistance program, AZ&Me Prescription Savings Program (AZ&Me), is part of the Company’s commitment to addressing barriers to access and affordability to improve medication adherence, enhance patient care, and help patients lead healthier lives. AZ&Me is just one of the ways that AstraZeneca makes its life-changing medicines widely available, accessible, and affordable.
For over 40 years, AstraZeneca has offered a patient assistance program through AZ&Me and prior legacy free drug programs, making it one of the longest standing patient assistance programs in the country. Since 2007, over five million people have benefited from this program. In addition to its patient assistance programs, AstraZeneca offers other affordability programs and resources to help increase patients’ access to medicines and reduce their out-of-pocket costs including a co-pay savings program for commercially-insured patients and additional affordability resources. Each of these programs offer financial support to particular patient populations, consistent with applicable legal requirements.
The goal of AZ&Me is to help patients who have been prescribed an AstraZeneca medication and are having difficulty affording it. Patients enrolled in AZ&Me receive their AstraZeneca medicine for free. To learn more, visit AZ&Me.com.
Global Initiative for Asthma. Updated May 2023. Accessed: March 2024. www.ginasthma.org
Price D, et al. Asthma control and management in 8,000 European patients: the REcognise Asthma and LInk to Symptoms and Experience (REALISE) survey. NPJ Prim Care Respir Med. 2014;24:14009.
Papi A, et al. Relationship of inhaled corticosteroid adherence to asthma exacerbations in patients with moderate-to-severe asthma. J Allergy Clin Immunol Pract. 2018;6(6): 1989-1998.e3.
Data on File. REF-173201. AstraZeneca Pharmaceuticals LP.
Sastre J, et al. Insights, attitudes, and perceptions about asthma and its treatment: a multinational survey of patients from Europe and Canada. World Allergy Organ J. 2016;9:13.
Fernandes AG, et al. Risk factors for death in patients with severe asthma. J Bras Pneumol. 2014;40(4):364-372.
Wark PA, et al. Asthma exacerbations. 3: Pathogenesis. Thorax. 2006;61(10):909-915.
Johnson DB, et al. Albuterol. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2024 Jan 10.
Montemayor T, et al. Albuterol: Often Used and Heavily Abused. Respiratory Care. November 2021, 66 (Suppl 10) 3603775.
Nwaru BI, et al. Overuse of short-acting β2-agonists in asthma is associated with increased risk of exacerbation and mortality: a nationwide cohort study of the global SABINA programme. Eur Respir J. 2020;55(4):1901872.
Lloyd A, et al. The impact of asthma exacerbations on health-related quality of life in moderate to severe asthma patients in the UK. Prim Care Respir J. 2007;16(1):22-27.
Bourdin A, et al. ERS/EAACI statement on severe exacerbations in asthma in adults: facts, priorities and key research questions. Eur Respir J. 2019;54(3):1900900.
Price DB, et al. Adverse outcomes from initiation of systemic corticosteroids for asthma: long-term observational study. J Asthma Allergy. 2018;11:193-204.
GOLD. Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2024. [Online]. Accessed: March 2024. goldcopd.org/2024-gold-report/
NHS hospitals have been hit by a UK-wide shortage of a life-saving drug used to keep alive patients who are at risk of dying because they cannot breathe without medical intervention.
Doctors have been told to ration their use of the liquid form of salbutamol, which plays a vital role in treating people suffering from severe asthma attacks or chronic obstructive pulmonary disease (COPD), which usually involves emphysema or chronic bronchitis.
A “safety critical” national patient safety alert issued by the Department of Health and Social Care (DHSC) and NHS England warns that 2.5mg and 5mg dose vials of salbutamol liquid are in short supply. The latter is “out of stock until mid-April 2024”.
The scarcity is so acute that hospitals were advised to “place urgent orders for unlicensed imports of salbutamol nebuliser liquid – do not wait for supplies to be exhausted before placing orders for imports”.
The drug is administered via a nebuliser, which pushes air through the liquid to create a mist that relaxes the patient’s muscles and reopens their airways.
The Guardian revealed in January that drug shortages in the UK were running at record levels, prompting fears among doctors that patients’ lives could be put at risk.
One specialist lung doctor who routinely uses nebules of the drug when patients can no longer breathe unaided said: “This is a worry. This is a life-saving drug that is the bread-and-butter medicine we use when patients with serious breathing problems are acutely unwell.
“We are being asked to ration it, and not to use it where possible and to use alternatives. We’ve been advised to use it sparingly – only if it’s absolutely essential. This isn’t a crisis at the moment. But it’s a worry that a life-saving drug is having to be rationed.”
The patient safety alert also told hospital bosses that in order to conserve supplies for use in the most serious cases, doctors should:
Wean all patients off nebulisers as soon as their condition has stabilised.
Consider no longer using nebuliser liquid for patients experiencing a mild to moderate asthma attack or flare-up of COPD and instead use a salbutamol pressurised metered-dose inhaler (pMDI).
When a patient does need nebuliser liquids, use them “when required rather than regularly”.
Supplies need to be used as far as possible only with “acute, severe exacerbations of COPD and asthma”, people who cannot breathe due to an attack of anaphylaxis – a life-threatening allergic reaction to eating something – and those who cannot use a pMDI.
The shortage does not affect the availability of salbutamol inhalers, the blue-coloured “reliever” inhalers that patients with lung conditions such as asthma use if they develop shortness of breath.
The scarcity only involves nebules, or vials, of liquid salbutamol, which is sold under various brand names including Ventolin. Patients are put on a salbutamol nebuliser when repeated inhalation of the drug through a tube has not helped them regain their capacity to breathe independently.
Doctors voiced concern about the situation. Dr Tim Cooksley, the immediate past president of the Society for Acute Medicine, said: “Salbutamol is commonly used to treat acutely unwell medical patients with breathing problems and there is not a ready alternative to it. It is an important part of daily practice and there is a risk of significant harm to these patients if supply issues are not resolved quickly.”
The charity Asthma and Lung UK posted a message on its website telling patients that “the supply of salbutamol nebuliser liquid is currently limited in the UK”.
It said: “Alternatives are available that healthcare professionals will be able to prescribe.” In addition, “nebuliser liquid from other countries that have similar high standards of licensing to the UK will also be made available.”
The DHSC said the shortage had come to an end after it arranged alternative supplies.
A spokesperson said: “Recent short-term disruption to the supply of salbutamol nebuliser liquid has now been resolved. This was caused by one supplier experiencing a manufacturing issue. The department quickly engaged with suppliers and others in the supply chain to ensure supplies were available for UK patients.”
Boehringer makes significant investment to improve the affordability of and access to its full range of inhaler products for COPD and asthma
Expansive program builds on the company's century-long commitment to patients with respiratory illnesses
RIDGEFIELD, Conn., March 7, 2024 /PRNewswire/ -- Boehringer Ingelheim today announced it will cap out-of-pocket costs at $35 per month for eligible patientsi for all the company's inhaler products. Boehringer's new program will dramatically decrease costs at the pharmacy counter for the most vulnerable patients, including those who are uninsured or underinsured. This reinforces the company's long-standing commitment to ensure access to important medicines for patients.
Boehringer Ingelheim 2024
"Patients have counted on Boehringer Ingelheim for nearly 140 years to tackle challenges across diseases, including respiratory illnesses," said Jean-Michel Boers, President and CEO, Boehringer Ingelheim USA Corporation. "The U.S. healthcare system is complex and often doesn't work for patients, especially the most vulnerable. While we can't fix the entire system alone, we are bringing forward a solution to make it fairer. We want to do our part to help patients living with COPD or asthma who struggle to pay for their medications. This new program supports patients with predictable, affordable costs at the pharmacy counter. We will also continue to advocate for substantive policy reforms to improve the healthcare system."
Starting June 1, 2024, eligible patients will pay no more than $35 a month at retail pharmacies for all Boehringer Ingelheim inhalers, including:
Boehringer's program builds on the company's long-standing commitment to supporting patients. The company will continue to provide access to free products for eligible patients and comprehensive patient support programs as well. In addition, Boehringer will decrease the list price on some of its inhaler products and will continue providing significant discounts and rebates off the list price of its medicines to insurers, pharmacy benefits managers and other parties, although unfortunately, these reductions are not always passed on to patients.
About Boehringer Ingelheim Boehringer Ingelheim is working on breakthrough therapies that transform lives, today and for generations to come. As a leading research-driven biopharmaceutical company, the company creates value through innovation in areas of high unmet medical need. Founded in 1885 and family-owned ever since, Boehringer Ingelheim takes a long-term, sustainable perspective. More than 53,000 employees serve over 130 markets in the two business units, Human Pharma and Animal Health. Learn more at www.boehringer-ingelheim.com/us/
i Terms and conditions apply. Government restrictions exclude people enrolled in federal government insurance programs from co-pay support.
ATROVENT HFA (ipratropium bromide HFA) Inhalation Aerosol is a prescription maintenance treatment for bronchospasm (airway narrowing) in patients with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema.
Important Safety Information
Do not use ATROVENT HFA if you are allergic to ipratropium or any of the ingredients in ATROVENT HFA or to atropine or similar drugs.
ATROVENT HFA is not a rescue medicine and should not be used for treating sudden breathing problems. Your doctor may give you other medicine to use for sudden breathing problems.
Allergic reactions may occur, including itching, swelling of the face, lips, tongue, or throat (involving difficulty in breathing or swallowing), rash, hives, bronchospasm (airway narrowing), or anaphylaxis. Some of these may be serious. If you experience any of these symptoms, stop taking ATROVENT HFA at once and call your doctor or get emergency help.
ATROVENT HFA can cause the narrowing of the airways to get worse (paradoxical bronchospasm) which may be life threatening. If this happens, stop taking ATROVENT HFA at once and call your doctor or get emergency help.
ATROVENT HFA may increase eye pressure which may cause or worsen some types of glaucoma. Do not get the spray into your eyes. The spray may cause eye pain or discomfort, blurred vision, enlarged pupils, seeing halos or colored images along with red eyes. If you have any of these symptoms, stop taking ATROVENT HFA and call your doctor right away.
Dizziness and blurred vision may occur with ATROVENT HFA. Should you experience these symptoms, use caution when engaging in activities such as driving a car or operating appliances or machinery.
ATROVENT HFA may cause difficulty with urination. Symptoms may include difficulty passing urine and/or painful urination. If you have any of these symptoms, stop taking ATROVENT HFA and call your doctor right away.
The most common side effects reported with use of ATROVENT HFA were bronchitis, COPD flare-up (exacerbation), shortness of breath and headache.
Tell your doctor about all medicines you are taking, including eye drops. Ask your doctor if you are taking any anticholinergic medicines because taking them together with ATROVENT HFA can increase side effects.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch or call 1‑800‑FDA‑1088.
(9/16) CL-AT-0017
Combivent Respimat (ipratropium bromide and albuterol) Inhalation Spray
Indication for Use
COMBIVENT RESPIMAT (ipratropium bromide and albuterol) is indicated for use in patients with chronic obstructive pulmonary disease (COPD) on a regular aerosol bronchodilator who continue to have evidence of bronchospasm (airway narrowing) and who require a second bronchodilator.
Important Safety Information
Do not use COMBIVENT RESPIMAT if you are allergic to any of the ingredients in COMBIVENT RESPIMAT or to atropine or other similar drugs.
COMBIVENT RESPIMAT can cause the narrowing of the airways to get worse (paradoxical bronchospasm) which may be life threatening. If this happens, stop taking COMBIVENT RESPIMAT at once and call your doctor or get emergency help.
COMBIVENT RESPIMAT can cause serious heart-related side effects, such as palpitations, chest pain, rapid heart rate, high blood pressure, tremor, or nervousness. Call your doctor if you experience any of these symptoms.
Avoid spraying COMBIVENT RESPIMAT into your eyes. COMBIVENT RESPIMAT may increase eye pressure which may cause or worsen some types of glaucoma. If you have sudden vision changes, eye pain or visual halos, stop taking COMBIVENT RESPIMAT and call your doctor right away.
COMBIVENT RESPIMAT may cause difficulty with urination.
Dizziness and blurred vision may occur with COMBIVENT RESPIMAT. Should you experience these symptoms, use caution when engaging in activities such as driving a car or operating appliances or other machines.
Do not use COMBIVENT RESPIMAT more often than your doctor has directed. Deaths have been reported with similar inhaled medicines in asthma patients who use the medicine too much. Seek medical attention if your treatment with COMBIVENT RESPIMAT becomes less effective for symptomatic relief, your symptoms become worse, and/or you need to use the product more frequently than usual.
Allergic reactions may occur, including itching, swelling of the face, lips, tongue, or throat (involving difficulty in breathing or swallowing), rash, hives, bronchospasm (airway narrowing), or anaphylaxis. Some of these may be serious. If you experience any of these symptoms, stop taking COMBIVENT RESPIMAT at once and call your doctor or get emergency help.
Tell your doctor about all your medical conditions, especially if you have narrow-angle glaucoma, prostate or urinary problems, a history of heart conditions (such as irregular heartbeat, high blood pressure), thyroid disorder, or diabetes. Also tell your doctor if you are pregnant or nursing. Tell your doctor about all medicines you are taking, especially heart medications or drugs to treat depression.
The most common side effects reported with use of COMBIVENT RESPIMAT include infection of the ears, nose, and throat, runny nose, cough, bronchitis, headache, and shortness of breath.
Click here for full Prescribing Information and Patient Instructions for Use.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch or call 1-800-FDA-1088.
SPIRIVA RESPIMAT, 2.5 mcg, and SPIRIVA HANDIHALER are long-term, once-daily, prescription maintenance medicines used to control symptoms of chronic obstructive pulmonary disease (COPD) by relaxing your airways and keeping them open. COPD includes chronic bronchitis and emphysema. SPIRIVA RESPIMAT and SPIRIVA HANDIHALER also reduce the likelihood of flare-ups (COPD exacerbations).
SPIRIVA RESPIMAT, 1.25 mcg, is a long-term, once-daily, prescription maintenance treatment of asthma for people 6 years and older.
SPIRIVA is not a treatment for sudden symptoms of asthma or COPD.
Important Safety Information for SPIRIVA RESPIMAT
Do not use SPIRIVA RESPIMAT or SPIRIVA HANDIHALER if you are allergic to tiotropium, ipratropium, atropine or similar drugs, or any ingredient in these medicines.
SPIRIVA RESPIMAT or SPIRIVA HANDIHALER are not rescue medicines and should not be used for treating sudden breathing problems. Your doctor may give you other medicine to use for sudden breathing problems.
SPIRIVA RESPIMAT or SPIRIVA HANDIHALER can cause allergic reactions. Symptoms can include raised red patches on your skin (hives), itching, rash and/or swelling of the lips, tongue, or throat that may cause difficulty in breathing or swallowing. If you have any of these symptoms, stop taking the medicine and seek emergency medical care.
Before using SPIRIVA HANDIHALER, tell your doctor if you have a severe allergy to milk proteins.
SPIRIVA RESPIMAT or SPIRIVA HANDIHALER can cause your breathing to suddenly get worse (bronchospasm). If this happens, use your rescue inhaler, stop taking SPIRIVA, and call your doctor right away or seek emergency medical care.
SPIRIVA RESPIMAT or SPIRIVA HANDIHALER can increase the pressure in your eyes (acute narrow-angle glaucoma) which can cause the following symptoms: eye pain, blurred vision, seeing halos or colored images along with red eyes. If you have any of these symptoms, stop taking your medicine and call your doctor right away.
Dizziness and blurred vision may occur with SPIRIVA RESPIMAT or SPIRIVA HANDIHALER. If you experience these symptoms, use caution when engaging in activities such as driving a car, or operating appliances or machinery.
SPIRIVA RESPIMAT or SPIRIVA HANDIHALER can cause new or worsened urinary retention. Symptoms of blockage in your bladder and/or enlarged prostate may include difficulty passing urine and/or painful urination. If you have any of these symptoms, stop taking your medicine and call your doctor right away.
The most common side effects reported with SPIRIVA RESPIMAT in patients with COPD include sore throat, cough, dry mouth, and sinus infection.
The most common side effects with SPIRIVA RESPIMAT in adult patients with asthma were sore throat, headache, bronchitis, and sinus infection. The side effect profile for adolescent and pediatric patients was comparable to that observed in adult patients with asthma.
The most common side effects reported with SPIRIVA HANDIHALER in patients with COPD include upper respiratory tract infection, dry mouth, sinus infection, sore throat, non-specific chest pain, urinary tract infection, indigestion, runny nose, constipation, increased heart rate, and blurred vision.
Do not swallow SPIRIVA capsules. The contents of the capsule should only be inhaled through your mouth using the HANDIHALER device.
Do not spray SPIRIVA RESPIMAT into your eyes, as this may cause blurring of vision and pupil dilation.
Tell your doctor about all your medical conditions including kidney problems, glaucoma, enlarged prostate, problems passing urine, or blockage in your bladder.
Tell your doctor all the medicines you take, including eye drops. Ask your doctor if you are taking any anticholinergic medicines because taking them together with SPIRIVA can increase side effects. Do not use SPIRIVA RESPIMAT and SPIRIVA HANDIHALER together.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.FDA.gov/medwatch or call 1-800-FDA-1088.
Click here for full Prescribing Information and Patient Instructions for Use for SPIRIVA RESPIMAT.
Click here for full Prescribing Information and Patient Instructions for Use for SPIRIVA HANDIHALER.
CL-SVR-0048 2.15.2017
STIOLTO RESPIMAT (tiotropium bromide and olodaterol) Inhalation Spray
APPROVED USE FOR STIOLTO RESPIMAT STIOLTO® RESPIMAT® (tiotropium bromide and olodaterol) Inhalation Spray is a prescription medicine used long term, 2 puffs 1 time each day, in controlling symptoms in adults with chronic obstructive pulmonary disease (COPD). COPD is a chronic lung disease that includes chronic bronchitis, emphysema, or both.
STIOLTO is not for treating sudden symptoms of COPD. Always have a rescue medicine with you to treat sudden symptoms.
STIOLTO is not for asthma.
Important Safety Information for STIOLTO RESPIMAT
Do not use STIOLTO if you have asthma. People with asthma who take long-acting beta2-agonist (LABA) medicines, such as olodaterol, (one of the medicines in STIOLTO), without also using a medicine called an inhaled corticosteroid, have an increased risk of serious problems from asthma, including being hospitalized, needing a tube placed in their airway to help them breathe, or death.
Do not use STIOLTO if you are allergic to tiotropium, ipratropium, atropine or similar drugs, olodaterol, or any ingredient in STIOLTO.
Call your healthcare provider or get emergency medical care if you experience symptoms of a serious allergic reaction including: rash, hives, itching, swelling of the face, lips, tongue, throat, and difficulties in breathing or swallowing.
Get emergency medical care if your breathing problems worsen quickly or if you use your rescue inhaler but it does not relieve your breathing problems. Call your healthcare provider if breathing problems worsen over time while using STIOLTO.
Do not use STIOLTO more often than prescribed by your doctor. Do not use STIOLTO with other LABAs or anticholinergics.
Do not use STIOLTO for treating sudden breathing problems. Always have a rescue inhaler with you to treat sudden symptoms.
Tell your doctor about all your medical conditions including heart problems, high blood pressure, seizures, thyroid problems, diabetes, kidney problems, glaucoma, enlarged prostate, and problems passing urine.
STIOLTO can cause serious side effects, including sudden shortness of breath that may be life threatening, fast or irregular heartbeat, increased blood pressure, chest pain, tremor, headache, nervousness, high blood sugar, or low blood potassium that may cause muscle weakness or abnormal heart rhythm. If any of these happens, stop taking STIOLTO and seek immediate medical help.
STIOLTO can cause new or worsening eye problems including narrow-angle glaucoma, and can increase the pressure in your eyes, which can cause the following symptoms: eye pain, blurred vision, seeing halos or colored images along with red eyes. If you have any of these symptoms, stop taking STIOLTO and call your doctor right away.
STIOLTO can cause new or worsened urinary retention. Symptoms of urinary retention may include difficulty passing urine, painful urination, urinating frequently, or urinating in a weak stream or drips. If you have any of these symptoms, stop taking STIOLTO and call your doctor right away.
The most common side effects of STIOLTO are runny nose, cough, and back pain.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, eye drops, vitamins, and herbal supplements. STIOLTO and certain other medicines may affect each other. STIOLTO is for oral inhalation only.
The STIOLTO cartridge is only intended for use with the STIOLTO RESPIMAT inhaler.
Do not spray STIOLTO into your eyes. Your vision may become blurred and your pupils may become larger (dilated).
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.FDA.gov/medwatchor call 1-800-FDA-1088.
Read the step-by-step patient Instructions for Use for STIOLTO RESPIMAT before you use your inhaler.
Striverdi® Respimat® (olodaterol) Inhalation Spray is a prescription medicine used long term, 2 puffs, 1 time each day, in controlling symptoms in adults with chronic obstructive pulmonary disease (COPD). COPD is a chronic lung disease that includes chronic bronchitis, emphysema, or both.
STRIVERDI RESPIMAT is not for treating sudden symptoms of COPD. Always have a rescue medicine with you to treat sudden symptoms. Striverdi is not for asthma.
Important Safety Information for STRIVERDI RESPIMAT
Do not use STRIVERDI if you have asthma. People with asthma who take long‐acting beta2‐agonist (LABA) medicines, such as STRIVERDI RESPIMAT, without also using a medicine called an inhaled corticosteroid (ICS), have an increased risk of serious problems from asthma, including being hospitalized, needing a tube placed in their airway to help them breathe, or death.
Get emergency medical care if your breathing problems worsen quickly or you use your rescue medicine but it does not relieve your breathing problems. Call your healthcare provider if breathing problems worsen over time while using STRIVERDI.
Do not use STRIVERDI RESPIMAT more often than prescribed for you by your doctor. Do not use STRIVERDI RESPIMAT with other LABAs.
Do not use STRIVERDI for treating sudden breathing problems. Always have a rescue inhaler with you to treat sudden symptoms.
Tell your doctor about all of your medical conditions, including heart problems, high blood pressure, seizures, thyroid problems, and diabetes.
STRIVERDI RESPIMAT can cause serious side effects including sudden shortness of breath that may be life threatening, fast or irregular heartbeat, increased blood pressure, chest pain, tremor, headache, nervousness, high blood sugar, or low blood potassium that may cause muscle weakness or abnormal heart rhythm. If any of these happen, stop taking STRIVERDI and seek immediate medical help.
Call your healthcare provider or get emergency medical care if you get any symptoms of a serious allergic reaction including: rash, hives, itching, swelling of the face, lips, tongue, throat, and difficulties in breathing or swallowing.
The most common side effects are runny nose, sore throat, upper respiratory tract infection, bronchitis, urinary tract infection, cough, dizziness, rash, diarrhea, back pain, and joint pain.
Tell your doctor about all the medicines you take, including prescription and non‐prescription medicines, vitamins, and herbal supplements. STRIVERDI and certain other medicines may affect each other.
STRIVERDI is for oral inhalation only.
The STRIVERDI cartridge is only intended for use with the STRIVERDI RESPIMAT inhaler.
Do not spray STRIVERDI into your eyes.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.FDA.gov/medwatch, or call 1‐800‐FDA‐1088.
Read the step-by-step patient Instructions for Use for STRIVERDI RESPIMAT before you use your inhaler.
Please see accompanying full Prescribing Information, including Patient Information, and Instructions for Use for STRIVERDI RESPIMAT.
CL-STRR-100001 6.5.2019
Media Contact: Michele Baer Human Pharma Communications [email protected]
The vaper or electronic cigarette has gained popularity in recent years as a healthy alternative to the traditional cigarette, especially among younger people. However, several studies have shown that vaping also has negative health effects. Discover the health problems caused by vaping and avoid it!
What is a vaper?
A vaper is a Electronic device which was born as a substitute for cigarettes. A device that heats an e-liquid until it turns into vapor so that it can be inhaled by the user. This vapor, in addition to nicotine, usually contains other compounds such as flavorings or cannabinoids, among others, which make its consumption harmful.
Health problems caused by vaping
Despite having gained popularity as a less harmful alternative to traditional cigarettes, several studies have shown that vaping also has negative health effects. And they are not few, nor unimportant. Today we analyze some of the most important ones so that you are aware of the dangers to which their consumption exposes you.
Addiction to nicotine and other substances
Vapers usually contain nicotine, a substance that It's addictive, especially when there is early exposure. Thus, consumed during adolescence, it can increase the risk of developing an addiction in the medium and long term. But vaper liquids without nicotine are not harmless either, as you will see, since they usually contain other substances, although less addictive and also not recommended.
Brain development
The health problems caused by vaping are greater among the youngest, among whom their consumption has skyrocketed in recent years. Yes, there are 12-year-old children vaping on devices provided by their own parents and that is a problem!
Young people's brains are especially susceptible to addiction because they are still developing. Furthermore, nicotine, beyond developing an addiction to these as we have already suggested, can negatively affect brain development, affecting memory, attention and learning.
Breathing difficulties
The liquids used in vapes contain numerous chemicals, including toxic compounds such as glycerin, propylene glycol, and formaldehyde, in addition to nicotine, of course. When these liquids are heated and subsequently inhaled, they can cause irritation of the lungs, which can result in respiratory problems such as cough, difficulty breathing, chronic bronchitis and even chemical pneumonia in the most serious cases.
In addition, the vapor released by vapes contains fine particles that enter the lungs, causing long-term inflammation. Numerous studies have proven that regular vaping can increase the risk of serious lung diseases such as pulmonary fibrosis.
One of the most harmful effects of vaping, therefore, is its impact on the respiratory system. It is especially dangerous in the people with asthma, since vaping can trigger an inflammatory response in the lungs and this in turn causes exacerbations of asthma symptoms, which in themselves are not few.
Cardiovascular diseases
In addition, vaping has also been linked in different studies to cardiovascular diseases. Some have shown that vaping can increase blood pressure and heart rate, which increases the risk of suffering a heart attack or stroke.
Sleep disturbance
Vaping also has a stimulating effect coming from its nicotine content. A fact that can make it difficult to fall asleep at bedtime and stay asleep at night. Therefore, vaping before bed is not recommended.
If you are thinking about starting to use a vaporizer to give up smoking, take into account the risks mentioned. It also ensures that this device, which is increasingly popular among adolescents, causes a dependency in them from which it is difficult to escape later. And if you have any questions about these devices or the health risks related to their use, do not hesitate to consult a medical professional for advice.
Chronic obstructive pulmonary disease (COPD) is a heterogeneous lung condition.1 A phenotype is generally considered to be the physical appearance or biochemical characteristic resulting from an interaction between your genotype and the environment. In COPD, where the underlying genes are mostly unknown or poorly characterized, phenotype has become almost synonymous with clinical subgroup.2
Phenotyping allows selecting a uniform group of patients and evaluating the most important outcome measures in this group for therapeutic clinical trials.3 The Spanish guide to chronic obstructive pulmonary disease (GEsEPOC) recognizes 3 phenotypes: emphysema, chronic bronchitis and asthma associated with COPD.4 The differences between the three phenotypes are not precisely known.
The forced oscillation technique (FOT), also referred to as respiratory oscillometry, is a non-invasive method able to provide a detailed analysis of the respiratory system resistance and reactance.5 This method has high potential to increase our understanding of the differences between phenotypes, as well as in their differential diagnosis.
Individuals with COPD exhibit multiple systemic manifestations, including a direct association between the decline in respiratory and peripheral muscle strength and their physical performance and overall functionality. The measurement of the respiratory pressures represent an important procedure for the functional evaluation of the respiratory muscles.6 In addition, peripheral muscle strength may be evaluated by the handgrip test. It is recognized for its cost-effectiveness, simplicity, and a robust correlation with morbidity in chronic diseases.7–11
Exercise intolerance is a common feature in patients with COPD, contributing to reduce the ability to perform activities of daily living.12 These abnormalities may be evaluated by the ADL-Glittre test, which proved to be valid, reliable and capable of reflecting the perception of functional limitation.13
In this context, the current study has two main objectives (1) use respiratory oscillometry to investigate the differences among the COPD phenotypes, and (2) evaluate the association between these abnormalities and the decrease in the functional performance of these patients.
Materials and Methods
Study Design
The present work was developed at the Biomedical Instrumentation Laboratory of the State University of Rio de Janeiro. This research is a cross-sectional study that was approved by the Ethics Committee of the Pedro Ernesto University Hospital (protocol 456 - CEP/2018/HUPE). All individuals signed an informed consent form before performing the tests. The study was carried out in accordance with the Declaration of Helsinki and all measurements were performed on the same day. The subjects carried out respiratory oscillometry and spirometry measurements before and after using the BD. Manovacuometry test, palmar grip and ADL–Glittre, were also performed, in that order.
Subjects
The number of volunteers was calculated using the MedCalc version 12 using preliminary results.14 It were assumed type I and type II errors of 5%, which are usual values in the literature. For the control group, individuals with normal spirometry, non-smokers, without previous pulmonary diseases, and with BMI within the normal range were included. Our study involved individuals who were diagnosed in accordance with the Global Initiative for Chronic Obstructive Lung Disease (GOLD)1 criteria and were aged 40 years or older. All studied subjects had no recent history of respiratory infections within the preceding thirty days at the time of the examinations, and they also had no past history of cardiovascular, orthopedic diseases or COVID-19.
The emphysema phenotype,4,15–17 the chronic bronchitis phenotype18 and the ACOS phenotype19 were diagnosed according to previous studies. Before conducting the tests, all patients continued their regular medications, excluding bronchodilators, in order to prevent any interference in the evaluation, as recommended by the American Thoracic Society/European Respiratory Society (ATS/ERS).20
Spirometry
For spirometry, a computerized system (nSpire Health, Inc., 1830 Left hand Circle, Longmont, CO 80501) was used according to standard protocols.20,21 The parameters analyzed were forced expiratory volume in one second (FEV1), forced vital capacity (FVC), FEV1/FVC ratio, and the ratio between forced expiratory flow (FEF) between 25% and 75% and FVC (FEF/FVC). These parameters were quantified in both absolute values and as a percentage of predicted values, with reference values derived from Pereira et al.22 Lung function data were acquired following post-bronchodilator testing.
Respiratory Oscillometry
The used instrument has been previously described23 and was employed in accordance with current recommendations.5 Pressure oscillations were applied in the frequency range of 4 to 32 Hz, with an amplitude of 2 cmH2O produced by a loudspeaker coupled to the respiratory system through a mouthpiece. The resulting flow and pressure signals were measured near the mouth by a pneumotachograph and a pressure transducer, respectively. During the exams, the volunteers remain seated, with their heads in a neutral position, use a nose clip, maintain spontaneous breathing through the mouthpiece and firmly supporting their cheeks and chin to minimize the shunt. A total of three acceptable tests, each comprising 16 seconds, were carried out, and the outcome considered was the average score. To eliminate any outlier values, only measurements with a coefficient of variation of respiratory resistance at the lowest frequency (4 Hz) equal to or less than 10% for all three tests were retained. Additionally, only examinations with a coherence function of 0.9 or greater across the entire frequency range were accepted, aiming to minimize the impact of spontaneous breathing.
The resistive properties were interpreted through the resistances at 4 Hz (R4), 12 Hz (R12), 20 Hz (R20) and the difference between R4 and R20 (R4-R20). The reactance results were interpreted using the mean (Xm), dynamic compliance (Cdyn), resonance frequency (fr) and area under the reactance curve (Ax). Cdyn is directly associated with the overall compliance of the respiratory system, and was computed using the reactance at 4 Hz (Cdyn=1/2πfX4). The resonance frequency, where respiratory reactance becomes zero, is an indicator of the homogeneity of the respiratory system. The parameter Ax was assessed by the area under the curve formed by the lowest frequency (4 Hz), the corresponding reactance (X4), and the resonance frequency (fr). To analyze the total mechanical load of the respiratory system, the impedance module at 4 Hz (Z4) was investigated, encompassing both the resistive and elastic components of the respiratory load.22
Manovacuometry
The maximum inspiratory pressure (MIP) and the maximum expiratory pressure (MEP) were measured. Measurements were performed five times, until three values were obtained with a variation of less than 5%, the highest value being considered for analysis. Predicted values were calculated using the formulas described in Black & Hyatt.6
Handgrip Test
The handgrip test was conducted using a handheld hydraulic dynamometer (Saehan, SH 5001). Participants were evaluated seated, with their elbows flexed at a 90° angle, holding the dynamometer in their hand in a neutral position. Three trials were performed with each hand, with a one-minute interval between measurements, and the highest value was used for analysis.24 Predicted values were derived from Novaes et al, 2009.25
ADL–Glittre
The ADL–Glittre test was performed as described in Skumlien et al 2006.26 Heart rate (HR), peripheral oxygen saturation (SpO2) and dyspnea index (Modified Borg Scale)27 were measured at the beginning, at each lap and at the end of the test. No verbal stimulus was offered throughout the test. The results obtained from patients with COPD were compared to reference values.28
Statistical Analysis
Data were initially tested for normality using the Shapiro–Wilk test (OriginLab® 8.0, Microcal Software, Inc. Ostend, Belgium), and when the sample showed a normal distribution behavior, the Two-Sample t-Test was used to analyze the groups. On the other hand, when the distribution presented a non-normal characteristic, the Mann–Whitney test was used to analyze the groups. The value of p < 0.05 was used to consider the statistically significant differences.
Correlation analyses were conducted using Pearson correlations for data that exhibited a normal distribution and Spearman correlations for data that did not adhere to a normal distribution. This analysis was carried out using Prism 5.03 (GraphPad Software, La Jolla CA, USA). The classification of these associations followed the guidelines proposed by Dawson and Trapp.29
The accuracy of oscillometry in distinguishing COPD phenotypes was assessed using receiver operating characteristic (ROC) analysis. Optimal prediction cut points were identified based on the optimal trade-off between specificity and sensitivity. The area under the curve (AUC) was computed to quantify the diagnostic accuracy, and AUC values greater than 0.80 were deemed suitable for diagnostic purposes.30 These results were presented as mean ± 95% of the confidence interval (CI). We evaluated oscillometry parameters pre and post bronchodilator, as well as the variations associated with the use of this drug (Δ=values post BD-values pre BD).
Results
The study included a cohort of 83 participants, comprising 20 control subjects and 63 patients with COPD (Table 1). Among these groups, no significant alterations were observed in terms of height, body mass, and body mass index (BMI). However, there was an increase in both age and pack-years within the COPD group.
Table 1 Biometric and Spirometric Parameters of the Studied Groups
Spirometric pre-bronchodilator (pre-BD) parameters exhibited significant reductions in individuals with COPD compared to the control group, as indicated in Table 1. Considering the BD effect, we observed a significantly higher change in ACOS in comparison with EMP and CB groups and that the EMP and CB groups presented similar modifications.
Oscillometric Parameters
Figure 1 depicts changes in resistive parameters. The values of R4 and R12 (Figures 1A and B, respectively) before BD use were significantly higher than those observed in the control group in all studied phenotypes. The use of the bronchodilator resulted in a significant reduction of R4 and R12 in patients with emphysema and ACOS, but not in patients with CB.
Figure 1 Resistive oscillometric parameters in patients classified according to the studied phenotypes. (A) R4, resistance at 4Hz; (B) R12, resistance at 12Hz; (C) R4-R20, resistance difference between 4 and 20Hz; ACOS, asthma COPD overlapping syndrome; The top and the bottom of the box plot represent the 25th- to 75th-percentile values while the circle represents the mean value, and the bar across the box represents the 50th-percentile value; ns, not significant; *p <0.05; **p <0.01; ***p<0.005; ****p<0.001.
R4-R20 values (Figure 1C) before BD use were significantly higher than those observed in the control group in all studied phenotypes. Bronchodilator use resulted in a significant reduction of R4-R20 in patients with ACOS, but not in patients with emphysema or CB.
Considering the comparisons among the phenotypes, higher values of R4 before BD were observed in the ACOS group in comparison with the group with emphysema (Figure 1A). The ACOS group showed significantly higher values of R4-R20 before using BD than that observed in EMP and CB groups (Figure 1C).
Figure 2A–D, shows that fr, Cdyn, Ax, and Z4 (respectively) values were significantly different from that observed in the control group before BD use. These parameters showed no observable differences following bronchodilator administration in patients with CB. In contrast, patients with emphysema and ACOS exhibited significant changes following bronchodilator administration.
Figure 2 Reactive oscillometric parameters in patients classified according to the studied phenotypes. (A) fr, resonance frequency; (B) Cdyn, dynamic complacency; (C) Ax, area under the reactance curve; (D) Z4, respiratory impedance module; (E) Xm, mean reactance; ACOS, asthma COPD overlapping syndrome; The top and the bottom of the box plot represent the 25th- to 75th-percentile values while the circle represents the mean value, and the bar across the box represents the 50th-percentile value; ns, not significant; *p <0.05; **p <0.01; ****p<0.001.
Xm values before BD use were more negative in all studied phenotypes than in the control group (Figure 2E). Bronchodilator administration resulted in significant increases in Xm in groups of patients with emphysema and ACOS. Patients with CB, however, do not present significant changes.
Functional Analysis Tests
Figure 3 describes predicted and measured values of handgrip test in each one of the studied subgroups of patients. Significant reductions were observed in all groups, both considering the dominant hand (Figure 3A) and the non-dominant hands (Figure 3B).
Figure 3 Predicted and measured handgrip values in the studied COPD phenotypes evaluated in the dominant (A) and non-dominant hands (B). ACOS, asthma COPD overlapping syndrome; The top and the bottom of the box plot represent the 25th- to 75th-percentile values while the circle represents the mean value, and the bar across the box represents the 50th-percentile value; ns, not significant; *p <0.05; **p <0.01; ***p<0.005; ****p<0.001.
Similar comparisons considering the respiratory pressures are showed in Figure 4. Significant changes were observed in all groups, both in MIP (Figure 4A) and MEP values (Figure 4B). Considering the comparisons among the phenotypes, higher values of MIP after BD were observed in the CB group in comparison with the group with emphysema (Figure 4A).
Figure 4 Predicted and measured maximum inspiratory pressure (MIP, (A) and maximum expiratory pressure (MEP, (B) values in the studied COPD phenotypes. ACOS, asthma COPD overlapping syndrome; ns, not significant; the top and the bottom of the box plot represent the 25th- to 75th-percentile values while the circle represents the mean value, and the bar across the box represents the 50th-percentile value; * p <0.05; ** p <0.01; **** p<0.001.
Figure 5 depicts the results of the AVD-Glittre test. The performed time significantly increased in all studied subgroups of patients when compared to predicted values.
Figure 5 Predicted and measured values of Glittre-ADL test in the studied COPD phenotypes. ACOS, asthma COPD overlapping syndrome; the top and the bottom of the box plot represent the 25th- to 75th-percentile values while the circle represents the mean value, and the bar across the box represents the 50th-percentile value; ns, not significant; ***p<0.005; ****p<0.001.
Correlation Analysis
Considering all COPD patients subgroups, almost all studied oscillometric parameters were associated with ADL-Glittre test time and handgrip analysis (Table 2). The exception was due to R4-R20. As can be seen in Table 2, no associations were observed among oscillometric parameters and respiratory pressures.
Table 2 Correlation Analysis Between Total Glittre-ADL Test Time, Handgrip Analysis, Respiratory Pressures and Oscillometric Parameters in the Whole Group of Patients with COPD
Considering only the emphysema phenotype (Table 3), Cdyn and Z4 showed significant inverse or direct associations, respectively (p < 0.05) with the ADL-Glittre test. There was no relationship between oscillometry and the palmar grip test in the dominant hand. With respect to the non-dominant hand, significant inverse correlations (p < 0.05) were observed between the resistive (R4) and reactive (fr) oscillometric parameters. There was no relationship between oscillometric parameters and manovacuometry.
Table 3 Correlation Analysis Between Total Glittre-ADL Test Time, Handgrip Analysis, Respiratory Pressures and Oscillometric Parameters in the Emphysema Group
When the correlation analysis included only CB patients, there was no relationship between the oscillometric parameters and the ADL-Glittre test (Table 4). No relationship was found with the palmar grip test in the dominant hand, and an inverse correlation was observed with R12 (p < 0.05). There was no relationship between oscillometric parameters and manovacuometry.
Table 4 Correlation Analysis Between Total Glittre-ADL Test Time, Handgrip Analysis, Respiratory Pressures and Oscillometric Parameters in the Chronic Bronchitis Group
Similar analysis considering only patients with the ACOS phenotype showed no relationship between the oscillometry and the ADL-Glittre test (Table 5). Concerning the palmar grip test, we do not observed associations with the dominant hand, while, significant inverse correlations (p < 0.05) were observed between R4 and R12 with the non-dominant hand. There was no relationship between oscillometric parameters and manovacuometry.
Table 5 Correlation Analysis Between Total Glittre-ADL Test Time, Handgrip Analysis, Respiratory Pressures and Oscillometric Parameters in the ACOS Group
Oscillometry Discriminating the Different Phenotypes
Oscillometric parameters pre and post bronchodilator do not present adequate values of AUC in discriminating the studied phenotypes (AUC < 0.80). The variations of R4-R20 due to the use of BD, on the other hand, provided an accurate discrimination of ACOS from emphysema (Figure 6A, AUC = 0.82, CI = 0.69±0.95) and chronic bronchitis (Figure 6B, AUC = 0.84, CI = 0.71±0.97).
Figure 6 Analysis of receiver operator characteristic (ROC) for the best parameter observed in the discrimination of ACOS from emphysema (A) and chronic bronchitis. (B) AUC, the area under the ROC curve; Δvariations of R4-R20 due to the use of bronchodilator; R4-R20, resistance at 4Hz minus at 20Hz.
Abbreviation: CI, confidence interval.
Discussion
In this study, four major findings were obtained: 1) initially, that oscillometry provided a detailed and consistent description of the COPD phenotypes; 2) BD response was different among the studied phenotypes; 3) The study revealed an association between oscillometry and functional capacity, especially within the emphysema phenotype; and 4) ROC curve analysis showed that ΔR4-R20 effectively discriminated ACOS from chronic bronchitis and emphysema phenotypes.
Table 1 displays the biometric parameters of the groups under investigation. Although there was a significant difference in age and body mass between the control group and the ACOS group, the analysed groups can be considered homogeneous, since height is the most important parameter for defining impedance values, age and BMI do not significantly alter respiratory oscillometry parameters.31 This parameter did not exhibit statistically significant differences among the studied groups (Table 1).
In general, the observed increases in R4 and R12 described in Figure 1A and B may be associated with inflammation of the mucous glands due to high tobacco consumption, which results in airway obstruction.32,33 Considering the specific characteristics of the phenotypes, the increased values in emphysema in comparison with the control group may be explained by the destruction of the small airways and loss of the parenchymal tissue that keeps the airways open.33 The increase in resistance found in the CB group may be associated with the worsening of airflow obstruction, a result of the excess mucus caused by the increase in goblet cells in the small airways in these individuals.34 The similar increase observed in ACOS can be explained by the typical increased bronchial secretion and, consequently, greater narrowing of the airways.
The comparisons among phenotypes showed that ACOS presented higher R4 values before BD use than the group with emphysema predominance. This is in line with the results of Van Noord et al, which showed that airway resistance was significantly higher in asthma and chronic bronchitis than in emphysema.35 Further support to this finding is provided by 3D CT analyses in the third to sixth generation central bronchus, which showed that the ACOS had greater airway narrowing compared with COPD.36
R4-R20 is associated with the respiratory system homogeneity of ventilation.37 The results of the present study provide evidence of reduced ventilation homogeneity in all studied phenotypes (Figure 1C). In close agreement with these results, Su et al, showed associations between variations in resistance and the degree of morphological abnormalities of the small airways evaluated with endobronchial optical coherence tomography in COPD and heavy smokers.38
R4-R20 increased in ACOS in comparison with emphysema and CB (Figure 1C). This result agrees with that obtained using 3D computed tomography analyses of the airways in COPD and ACOS in the study of Karayama et al.36 This difference may be explained, at least in part, by the addition of the pathophysiological characteristics of asthma in these patients. In this disease, bronchial obstruction may result from bronchospasm, mucosal oedema and hypersecretion.39 Thus, the presence of these additional factors seems to introduce greater non-homogeneities in the ventilation of these patients than those caused in patients with predominance of emphysema or CB.
Some authors claim that bronchodilation in patients with COPD causes an increase in the diameter of the airways.40,41 In line with previous studies,41,42 we found a reduction in respiratory resistance (Figures 1A and B) in individuals classified as having emphysema and ACOS after using a bronchodilator. It was interesting to observe that similar alterations did not occur in the group with a preponderance of CB. This indicates that the smooth muscle relaxation introduced by BD use do not result in significant changes in this group. Since the main mechanism of respiratory obstruction in these patients refers to excessive mucus production, which is not influenced by BD use, this result seems to be reasonable. Important structural changes in CB includes airway wall thickening due to remodeling. This phenomena make airways difficult to “open”, reducing response to bronchodilator agents.43 Baldi et al suggested that airway distensibility is reduced in COPD and that airway smooth muscle contributes to the increased airway stiffness in COPD subjects with prevailing bronchitis,44 but not in those with more emphysema.
Bronchodilator use decreased the value of R4-R20 in patients with emphysema and ACOS (Figure 1C), revealing an improvement in the ventilation homogeneity.45 Although R4-R20 was reduced with the BD use, this parameter still presented increased values compared to healthy individuals. This indicates that not all imbalances in the time constants were eliminated with the BD use.46 In a similar way to what was observed in R4 (Figure 1A) and R20 (Figure 1B), the R4-R20 values did not change with the use of the bronchodilator. We can speculate that the same reasons described earlier for the lack of response in terms of R4 and R20 may also be involved in these results.43,44
A comparative analysis of the reactive parameters before BD use with the control group showed significant changes in all studied parameters and phenotypes (Figure 2). This can be explained by the increased ventilation non-homogeneity in the respiratory system of these patients, which occurs due to the increase in imbalances in the time constants of the different regions of the lungs of patients with COPD.47 These changes may also be associated with abnormalities in lung tissue, chest wall, airway distensibility and increased resistance,47 as well as with the reduction in the apparent compliance.48
The use of bronchodilator medication significantly improved almost all reactive parameters in the emphysema and ACOS groups (Figure 2). The bronchodilator use relax the smooth muscles of the bronchi, improving the compliance of the airway wall.49 A previous study showed an increase in Cdyn after the use of salbutamol in patients with obstruction due to asthma and COPD.50 The increase in compliance reflects the improvement in lung expansion, associated with a reduction in the peripheral airways resistance resulting in an improvement in lung homogeneity and a decrease in hyperinflation after drug inhalation.50 The cited factors explain the improvements observed in Cdyn, AX, Z4 and Xm (Figure 2).
In contrast, the use of BD did not result in discernible changes in patients with CB (Figure 2). Although persistent airflow limitation occurs in patients with COPD, ACOS and asthma, the flow limitation phenomena may have distinct characteristics, resulting in different responsiveness to bronchodilators.51 Chronic bronchitis is caused by the hypersecretion of mucus by the goblet cells, which leads to the worsening of resistance to airflow by obstructing the lumen of the small airways. The presence of inflammation in the epithelium of the central airways is another important characteristic, which may introduce epithelial remodelling.52,53 On the other hand, the main characteristic of emphysema refers to the destruction of the lung parenchyma, leading to loss of elastic recoil.54 Previous works hypothesize that an inflamed and thick airway may appear more rigid than an airway subjected to the devastation of proteases.55,56 This factor could explain, at least in part, observed differences between CB and emphysema, since relaxation in the airways smooth muscles could have lesser effects in more rigid airways, such as those present in CB than in the airways of patients with emphysema.
Asthma is characterized by bronchial muscle contraction and airway narrowing that is highly reversible using BD medication.35 The greater response to BD observed in patients with ACOS compared to those with CB (Figure 2) is probably related to this characteristic.
Respiratory abnormalities represent only one aspect of the multifaceted complications associated with this COPD. In this context, muscle dysfunction emerges as a primary anomaly linked to diminished functionality.57 The evaluation of peripheral muscle strength in COPD patients carries paramount importance due to its correlation with various factors, including exercise intolerance, challenges in performing daily activities, and a diminished quality of life.57 This study observed substantial reductions in manual grip strength for both the dominant (Figure 3A) and non-dominant hands (Figure 3B) within the emphysema, chronic bronchitis, and ACOS groups when contrasted with predicted values for each respective group. The etiology of muscle dysfunction and exercise impediments observed in COPD patients is explicable through systemic inflammation originating from the pulmonary system and a reduction in the bulk of musculature within the lower extremities.10 Musculoskeletal dysfunction, characterized by the loss of muscle strength and endurance, is principally attributed to diminished muscle area, reduced lean body mass, compromised muscle stamina, and an augmented susceptibility to fatigue.58,59 Holden et al described a relation between reduced handgrip strength and diminished quality of life, heightened vulnerability to exacerbated COPD morbidity and increased risk of mortality.58
Significant reductions in manometry were observed for PiMax (Figure 4A) and PeMax (Figure 4B) in the emphysema, chronic bronchitis, and ACOS groups. Muscle fatigue resulting from the detrimental effects of COPD not only affects peripheral muscles but can also compromise respiratory muscle function.60 Another explanation is associated with airway obstruction and pulmonary hyperinflation, which position the diaphragm at a mechanical disadvantage, resulting in a chronic decrease in contact area, leading to reduced respiratory muscle efficiency in these individuals.61
Significant increases in the time taken to complete the Glittre-ADL test were observed in the emphysema, chronic bronchitis, and ACOS groups compared to predicted values (Figure 5). These limitations are associated with several factors, including gas exchange inefficiency, ventilatory limitation, peripheral muscle weakness, alterations in metabolism, and peripheral muscle composition.62 Dynamic hyperinflation may contribute to these findings, potentially worsening when respiratory demand increases during exercise and creating a sensation of dyspnea as respiratory work intensifies.63
Table 2 shows that functional capacity was more sensitive to changes in airway obstruction (R4 and R12) and elastic properties (fr, Cdyn, Ax and Z4) than changes in ventilation heterogeneity (R4-R20). The moderate to good correlations observed among oscillometry with ADL-Glittre test and Handgrip analysis reinforce the notion that oscillatory indices are associated with physical performance and are valuable for predicting reduced exercise tolerance in individuals with COPD.64 The results are also consistent with that obtained recently using the 6-minute walking distance65,66 and during cycle ergometer tests.67 These associations agree with the involved physiology, describing an increase in ADL-Glittre test time with airway obstruction and Cdyn reduction. This reflects the systemic effects due to lung abnormalities, including the presence of airflow limitation and dynamic hyperinflation.
Considering the analysis performed specifically in the studied COPD phenotypes, it was interesting to note significant relationships in the emphysema group (Table 3) describing a decrease in ADL-Glittre test performance with reductions in Cdyn and increases in Z4. These associations agree with the typical changes observed in COPD, describing abnormalities in elastic properties (Cdyn), and respiratory work (Z4). The inverse associations observed among resistive properties (R4) and ventilation heterogeneity (fr) with Handgrip analysis are also consistent with the cited principles.
A recent review points out that oscillometry adds insight into the pathophysiology of COPD, and that we still need more data to assess how this method relates to clinical phenotypes of COPD.37 COPD phenotypes should be able to classify patients into distinct subgroups that provide prognostic information and allow us to better determine the appropriate therapy that alters clinically significant outcomes.3,37 In this sense, the current study provides evidence that oscillometry may help to accurately discriminate ACOS from emphysema and chronic bronchitis (AUC>0.80). This high performance probably reflects the higher impact of bronchodilator use in the ventilation heterogeneity of the ACOS group due to the asthmatic component of this phenotype.
The differential diagnosis between groups of emphysema predominance and chronic bronchitis predominance are likely to be complex, and their clarification needs further investigation. It is important to emphasize that promising values of AUC were observed in these analyses, with values around 0.65. Previous studies from our research group have demonstrated that the application of artificial intelligence methods can enhance the accuracy of oscillometry parameters in the early diagnosis of smoking-induced respiratory changes68 and in the diagnosis69 and classification70 of COPD. In this manner, the application of these methods for the proper discrimination between emphysema and chronic bronchitis is one of the upcoming steps planned by our research group in this line of investigation.
A thorough examination of potential limitations of the current study is necessary. Firstly, the study concentrated on whole-breath impedance measurements and did not evaluate within-breath analysis.71 In future research, it is advisable to explore similar analyses focusing on within-breath impedance parameters. This avenue represents a promising research direction. Another noteworthy point; plethismographic exams were not feasible in all patients due to their clinical condition or inability to cooperate during the test. This resulted in the loss of important information that could have helped to clarify the differences between the phenotypes. Lastly, this is a single-center study, thus the outcomes may lack broad applicability to the entire patient demographic. This emphasizes the necessity for future investigations with a higher number of volunteers. Despite these limitations, this preliminary analysis significantly contributes to a critical discussion concerning the use of oscillometric parameters to evaluate COPD phenotypes.
Conclusion
In conclusion, this study set out to examine COPD phenotypes in-depth via respiratory oscillometry. It has been shown initially that oscillometry provided a description of the COPD phenotypes consistent with the involved physiopathology. The use of BD medication introduced clear changes in ACOS and Emphysema, but not in patients with predominance of CB. The correlation analysis unveiled a clear relationship between oscillometry and functional capacity, notably within the emphysema phenotype. ROC analysis further indicated that oscillometric parameters displayed sufficient accuracy in discriminating ACOS from Emphysema and CB. These findings offer evidence that oscillatory indices have the potential to enhance our understanding and identification of COPD phenotypes.
Acknowledgments
The research presented in this study received support from the Brazilian Council for Scientific and Technological Development (CNPq), the Rio de Janeiro State Research Supporting Foundation (FAPERJ), and was partially funded by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brasil (CAPES) under Finance Code 001.
Disclosure
The authors report no conflicts of interest in this work.
References
1. The global strategy for the diagnosis, management and prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD); 2023. Available from: wwwgoldcopdorg. Accessed February 29, 2024.
2. Vestbo J. COPD: definition and Phenotypes. Clinics Chest Med. 2014;35(1):1–6. doi:10.1016/j.ccm.2013.10.010
3. Han M, Agusti A, Calverley P, et al. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med. 2010;182(5):598–604. doi:10.1164/rccm.200912-1843CC
4. Miravitlles M, Soler-Cataluña JJ, Calle M, et al. Spanish COPD Guidelines (GesEPOC) 2017. Pharmacological treatment of stable chronic obstructive pulmonary disease. Archivos de Bronconeumología. 2017;53(6):324–335. doi:10.1016/j.arbres.2017.03.018
5. King G, Bates J, Berger K, et al. Technical standards for respiratory oscillometry. Europ resp J. 2020;55(2):1900753. doi:10.1183/13993003.00753-2019
6. Black LF, Hyatt RE. Maximal respiratory pressures: normal values and relationship to age and sex. Am Rev Respir Dis. 1969;99(5):696–702. doi:10.1164/arrd.1969.99.5.696
7. Marcos L, Bichinho GL, Panizzi EA, Storino KKG, Pinto DV. Analysis of chest radiography of individuals with COPD and its correlation with functional testing. Fisioterapia e Movimento. 2012;25:629–637. doi:10.1590/S0103-51502012000300018
8. Cheung CL, Nguyen US, Au E, Tan KC, Kung AW. Association of handgrip strength with chronic diseases and multimorbidity: a cross-sectional study. Age. 2013;35(3):929–941. doi:10.1007/s11357-012-9385-y
9. Strandkvist V, Backman H, Röding J, Stridsman C, Lindberg A. Hand grip strength is associated with forced expiratory volume in 1 second among subjects with COPD: report from a population-based cohort study. Int J Chron Obstruct Pulmon Dis. 2016;11:2527–2534. doi:10.2147/COPD.S114154
10. Jeong M, Kang H, Song P, et al. Hand grip strength in patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2017;12:2385–2390. doi:10.2147/COPD.S140915
11. Albarrati A, Gale N, Enright S, Munnery M, Cockcroft J, Shale D. A simple and rapid test of physical performance in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2016;11:1785–1791. doi:10.2147/COPD.S106151
12. Karloh M, Karsten M, Pissaia F, Araujo C, Mayer A. Physiological responses to the Glittre-ADL test in patients with chronic obstructive pulmonary disease. J Rehabil Med. 2014;46(1):88–94. doi:10.2340/16501977-1217
13. Gulart A, Munari A, Klein S, Santos da Silveira L, Mayer A. The glittre-ADL Test cut-off point to discriminate abnormal functional capacity in patients with COPD. COPD. 2018;15(1):73–78. doi:10.1080/15412555.2017.1369505
14. Teixeira EM, Lopes AJ, Melo PL Differences in respiratory mechanics in emphysema and chronic bronchitis evaluated by forced oscillations. In: XXVII Brazilian Congress on Biomedical Engineering: Proceedings of CBEB 2020. Vitória, Brazil Cham: Springer International Publishing; 2022:285–291.
15. Izquierdo-Alonso JL, Rodriguez-Gonzalezmoro JM, De lucas-ramos P, et al. Prevalence and characteristics of three clinical phenotypes of chronic obstructive pulmonary disease (COPD). Respir Med. 2013;107(5):724–731. doi:10.1016/j.rmed.2013.01.001
16. Golpe R, Suárez-Valor M, Martín-Robles I, et al. Mortality in COPD patients according to clinical phenotypes. Int J Chron Obstruct Pulmon Dis. 2018;13:1433–1439. doi:10.2147/COPD.S159834
17. Cheng Y, Tu X, Pan L, et al. Clinical characteristics of chronic bronchitic, emphysematous and ACOS phenotypes in COPD patients with frequent exacerbations. Int J Chron Obstruct Pulmon Dis. 2017;12:2069–2074. doi:10.2147/COPD.S140231
18. Koblizek V, Chlumsky J, Zindr V, et al. Chronic Obstructive Pulmonary Disease: official diagnosis and treatment guidelines of the Czech Pneumological and Phthisiological Society; a novel phenotypic approach to COPD with patient-oriented care. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2013;157(2):189–201. doi:10.5507/bp.2013.039
19. Soler-Cataluña JJ, Cosío B, Izquierdo JL, et al. Consensus document on the overlap phenotype COPD-asthma in COPD. Arch Bronconeumol. 2012;48(9):331–337. doi:10.1016/j.arbres.2011.12.009
20. Miller M, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. doi:10.1183/09031936.05.00034805
21. Graham B, Steenbruggen I, Miller M, et al. Standardization of spirometry 2019 update. an official American thoracic society and European respiratory society technical statement. Am J Respir Crit Care Med. 2019;200(8):e70–e88. doi:10.1164/rccm.201908-1590ST
22. Pereira C, Sato T, Rodrigues S. New reference values for forced spirometry in white adults in Brazil. J Bras Pneumol. 2007;33(4):397–406. doi:10.1590/S1806-37132007000400008
23. de Melo PL, Werneck MM, Giannella-Neto A. New impedance spectrometer for scientific and clinical studies of the respiratory system. Rev Sci Instrum. 2000;71(7):2867–2872. doi:10.1063/1.1150705
24. Mathiowetz V, Weber K, Volland G, Kashman N. Reliability and validity of grip and pinch strength evaluations. J Hand Surg. 1984;9(2):222–226. doi:10.1016/S0363-5023(84)80146-X
25. Novaes RD, Miranda AS, Silva JO, Tavares BVF, Dourado VZ. Reference equations for predicting of handgrip strength in Brazilian middle-aged and elderly subjects. Fisioter Pesq. 2009;16(3):217–222. doi:10.1590/S1809-29502009000300005
26. Skumlien S, Hagelund T, Bjortuft O, Ryg M. A field test of functional status as performance of activities of daily living in COPD patients. Respir Med. 2006;100(2):316–323. doi:10.1016/j.rmed.2005.04.022
27. Borg GA. Psychophysical bases of perceived exertion. Med Sci Sports Exercise. 1982;14(5):377???381. doi:10.1249/00005768-198205000-00012
28. Reis C, Karloh M, Fonseca F, Biscaro R, Mazo G, Mayer A. Functional capacity measurement: reference equations for the Glittre Activities of Daily Living test. J Bras Pneumol. 2018;44(5):370–377. doi:10.1590/s1806-37562017000000118
30. Swets JA. Measuring the accuracy of diagnostic systems. Science. 1988;240(4857):1285–1293. doi:10.1126/science.3287615
31. Ribeiro F, Lopes A, Melo P. Reference values for respiratory impedance measured by the forced oscillation technique in adult men and women. Clin Respir J. 2018;12(6):2126–2135. doi:10.1111/crj.12783
32. Bohadana A, Teculescu D, Martinet Y. Mechanisms of chronic airway obstruction in smokers. Respir Med. 2004;98(2):139–151. doi:10.1016/j.rmed.2003.09.005
33. Loring SH, Garcia-Jacques M, Malhotra A. Pulmonary characteristics in COPD and mechanisms of increased work of breathing. J Appl Physiol. 2009;107(1):309–314. doi:10.1152/japplphysiol.00008.2009
34. Widysanto A, Mathew G. Chronic Bronchitis. In: StatPearls. StatPearls Publishing; 2021.
35. Van Noord J, Clement J, Van de Woestijne K, Demedts M, Taussig LM. Total respiratory resistance and reactance in patients with asthma, chronic bronchitis, and emphysema. J Am Rev Respir Dis. 1991;143(5 Pt 1):922–927. doi:10.1164/ajrccm/143.2.312
36. Karayama M, Inui N, Yasui H, et al. Physiological and morphological differences of airways between COPD and asthma–COPD overlap. J Sci Rep. 2019;9(1):7818. doi:10.1038/s41598-019-44345-6
37. Kaminsky D, Simpson S, Berger K, et al. Clinical significance and applications of oscillometry. Eur Respir Rev. 2022;31(163):210208. doi:10.1183/16000617.0208-2021
38. Su Z-Q, Guan W-J, S-Y L, et al. Significances of spirometry and impulse oscillometry for detecting small airway disorders assessed with endobronchial optical coherence tomography in COPD. J Int J Chron Obstruct Pulm Dis. 2018;Volume 13:3031–3044. doi:10.2147/COPD.S172639
39. Spiro SG, Silvestri GA, Agustí A. Clinical respiratory medicine e-book: expert consult-online and print. Elsevier Health Sci. 2012;2:1.
40. Borrill ZL, Houghton C, Woodcock A, Vestbo J, Singh D. Measuring bronchodilation in COPD clinical trials. J Br Clin Pharmacol. 2005;59(4):379–384. doi:10.1111/j.1365-2125.2004.02261.x
41. da Costa GM, Faria AC, Di mango AM, Lopes AJ, Lopes de Melo P. Respiratory impedance and response to salbutamol in healthy individuals and patients with COPD. Respiration. 2014;88(2):101–111. doi:10.1159/000362691
42. Costa G, Faria A, Mango A, Lopes A, Jansen J, Melo P. Bronchodilation in COPD: beyond FEV1-The effect of albuterol on resistive and reactive properties of the respiratory system. J Bras Pneumol. 2009;35(1):1. doi:10.1590/s1806-37132009000100001
43. Jones RL, Noble PB, Elliot JG, James AL. Airway remodelling in COPD: it’s not asthma! Respirology. 2016;21(8):1347–1356. doi:10.1111/resp.12841
44. Baldi S, Dellacà R, Govoni L, et al. Airway distensibility and volume recruitment with lung inflation in COPD. J Appl Physiol. 2010;109(4):1019–1026. doi:10.1152/japplphysiol.00147.2010
45. Zerah F, Lorino A-M, Lorino H, Harf A, Macquin-Mavier I. Forced oscillation technique vs spirometry to assess bronchodilatation in patients with asthma and COPD. J Chest. 1995;108(1):41–47. doi:10.1378/chest.108.1.41
46. Costa GM, Faria ACD, Mango D, et al. Broncodilatação na DPOC: muito além do VEF1-efeito do salbutamol nas propriedades resistivas e reativas do sistema respiratório. J Brasil de Pneumol. 2009;35(4):325–333. doi:10.1590/S1806-37132009000400006
47. Di Mango A, Lopes A, Jansen J, Melo P. Changes in respiratory mechanics with increasing degrees of airway obstruction in COPD: detection by forced oscillation technique. Respir Med. 2006;100(3):399–410. doi:10.1016/j.rmed.2005.07.005
48. Faria ACD, da Costa AA, Lopes AJ, Jansen JM, de Melo PL. Forced oscillation technique in the detection of smoking‐induced respiratory alterations: diagnostic accuracy and comparison with spirometry. Clinics. 2010;65(12):1295–1304. doi:10.1590/S1807-59322010001200012
49. Delacourt C, Lorino H, Herve-Guillot M, Reinert P, Harf A, Housset B. Use of the Forced Oscillation Technique to Assess AirwayObstruction and Reversibility in Children. American Journal of Respiratory and Critical Care Medicine. 2000;161(3):730–736. doi:10.1164/ajrccm.161.3.9904081
50. Lorino A, Zerah F, Mariette C, Harf A, Lorino H. Respiratory resistive impedance in obstructive patients: linear regression analysis vs viscoelastic modelling. Europ resp J. 1997;10(1):150–155. doi:10.1183/09031936.97.10010150
51. Kitaguchi Y, Yasuo M, Hanaoka M. Comparison of pulmonary function in patients with COPD, asthma-COPD overlap syndrome, and asthma with airflow limitation. J Int J Chron Obstruct Pulm Dis. 2016;991–997. doi:10.2147/COPD.S105988
52. Kim V, Criner GJ. Chronic bronchitis and chronic obstructive pulmonary disease. J Am J Respir Crit Care Med. 2013;187(3):228–237. doi:10.1164/rccm.201210-1843CI
53. West J, Luks A. West’s Pulmonary Pathophysiology: The Essentials. 10 ed. Wolters Kluwer; 2022.
54. Márquez-Martín E, Ramos PC, López-Campos JL, et al. Components of physical capacity in patients with chronic obstructive pulmonary disease: relationship with phenotypic expression. J Int J Chron Obstruct Pulm Dis;2011. 105–112. doi:10.2147/COPD.S16646
55. Bates JH. Systems physiology of the airways in health and obstructive pulmonary disease. J Wiley Interdiscipl Rev. 2016;8(5):423–437.
56. Khan MA, Ellis R, Inman MD, Bates JH, Sanderson MJ, Janssen LJ. Influence of airway wall stiffness and parenchymal tethering on the dynamics of bronchoconstriction. Am J Physiol Lung Cell Mole Physiol. 2010;299(1):L98–L108. doi:10.1152/ajplung.00011.2010
57. Nunes MF, Hervé BB, Lukrafka JL, Monteiro MB. Handgrip strength and its relation to isokinetic dynamometry in COPD. J Fisioterapia Em Movime. 2020;3:33.
58. Holden M, Fyfe M, Poulin C, et al. Handgrip strength in people with chronic obstructive pulmonary disease: a systematic review and meta-analysis. J Phys Ther. 2021;101:6.
59. Vieira RHG, Nogueira IDB, Queiroz NF, et al. Peripheral and respiratory muscle strength in chronic obstructive pulmonary disease. Rev bras cineantropom desempenho hum. 2018;20(2):125–133. doi:10.5007/1980-0037.2018v20n2p125
60. Souza RM, Cardim AB, Maia TO, Rocha LG, Bezerra SD, Marinho PM. Inspiratory muscle strength, diaphragmatic mobility, and body composition in chronic obstructive pulmonary disease. J Physiother Res Int. 2019;24:2.
61. Dellacà RL, Santus P, Aliverti A, et al. Detection of expiratory flow limitation in COPD using the forced oscillation technique. Eur Respir J. 2004;23(2):232–240. doi:10.1183/09031936.04.00046804
62. Corrêa KS, Karloh M, Martins LQ, Santos K, Mayer AF. Can the Glittre ADL test differentiate the functional capacity of COPD patients from that of healthy subjects? J Braz J Phy Ther. 2011;15(6):467–473. doi:10.1590/S1413-35552011005000034
63. Raskin J, Marks T, Miller A. Phenotypes and Characterization of COPD. J Cardiopul Rehabil Prevent. 2018;38(1):43–48. doi:10.1097/HCR.0000000000000271
64. Ribeiro CO, Lopes AJ, de Melo PL. Respiratory oscillometry in chronic obstructive pulmonary disease: association with functional capacity as evaluated by adl glittre test and hand grip strength test. Int J Chronic Obstr. 2022;Volume 17:1017–1030. doi:10.2147/COPD.S353912
65. Yamamoto A, Shirai T, Hirai K, et al. Oscillometry as a predictor of exercise tolerance in COPD. Copd. 2020;17(6):647–654. doi:10.1080/15412555.2020.1844176
66. Zeng G-S, Chen L-C, Fan H-Z, et al. The relationship between steps of 6MWT and COPD severity: a cross-sectional study. Int J Chronic Obstr. 2018;14:141–148.
67. Yamamoto Y, Miki K, Matsuki T, et al. Evaluation of exertional ventilatory parameters using oscillometry in COPD. Int J Chron Obstruct Pulmon Dis. 2020;15:1697–1711. doi:10.2147/COPD.S260735
68. Amaral JL, Lopes AJ, Jansen JM, Faria AC, Melo PL. An improved method of early diagnosis of smoking-induced respiratory changes using machine learning algorithms. Comput Methods Programs Biomed. 2013;112(3):441–454. doi:10.1016/j.cmpb.2013.08.004
69. Amaral JL, Lopes AJ, Jansen JM, Faria AC, Melo PL. Machine learning algorithms and forced oscillation measurements applied to the automatic identification of chronic obstructive pulmonary disease. Comput Methods Programs Biomed. 2012;105(3):183–193. doi:10.1016/j.cmpb.2011.09.009
70. Amaral JL, Lopes AJ, Faria AC, Melo PL. Machine learning algorithms and forced oscillation measurements to categorise the airway obstruction severity in chronic obstructive pulmonary disease. Comput Methods Programs Biomed. 2015;118(2):186–197. doi:10.1016/j.cmpb.2014.11.002
71. András L, Dorottya C, Zoltán G, et al. Airway dynamics in COPD patients by within-breath impedance tracking: effects of continuous positive airway pressure. Eur Respir J. 2017;49(2):1601270. doi:10.1183/13993003.01270-2016
Doctors in the UAE are highlighting at least a 10 per cent surge in patients seeking medical attention for persistent coughs.
They said weather fluctuations commonly act as triggers for conditions such as asthma, allergies, and bronchitis, leading to an escalation in chronic cough during these changes.
Medics explained chronic cough often stems from respiratory issues like asthma, Chronic obstructive pulmonary disease (COPD), pulmonary fibrosis, bronchiectasis, and various other respiratory causes.
Dr Jimmy Joseph, Specialist Internal Medicine and Diabetologist, Aster Clinic, International City said, “There is a surge in cases of cough. I get to see 8-10 patients daily in OPD with disturbing coughs lasting more than 10 days. We see persistent coughs greater than three weeks with nearly 20-30 per cent of daily cases. Persistent cough means when the cough lasts between three to eight weeks.”
Dr Jimmy Joseph
Acid reflux-induced cough
Even Gastroesophageal reflux disease (GERD) is a condition where stomach acid regularly flows back into the esophagus, irritating the lining. When this acidic fluid reaches the throat and respiratory tract, it can lead to irritation and trigger a cough.
“Causes include post viral/ post-infective cough, postnasal drip, GERD/ acid reflux, asthma, and smoking. Other causes include chronic bronchitis/COPD, Covid 19 and post-infection, ACE inhibitors (blood pressure medication), congestive heart failure, and lung cancer,” he added.
Medics stressed the substantial increase in cough cases can be attributed significantly to the changing seasons, the flu, influenza, cold weather, rain, and dust.
“Patients should approach a doctor when a cough lasts more than 7-10 days, a person loses weight rapidly, coughs out blood, has continuous fever, night sweats, chest pain, and shortness of breath. If your doctor prescribes an antibiotic, complete the full antibiotic course. Avoid OTC medications,” Joseph added.
Dr Bassam Abdelmonem, consultant Emergency Care with Prime Hospital, also reiterated that they’ve recently observed an increased number of patients with chronic coughs.
Dr Bassam Abdelmonem
He said, “Around 10 per cent of patients visiting the Emergency Room (ER) present themselves with chronic cough. Weather changes are common asthma triggers; allergies and bronchitis then chronic cough will increase by these changes. Patients with chronic cough should seek medical advice when they have had a cough for more than three weeks persistently or they're losing weight for no reason. Other reasons include if one has a weakened immune system – for example, because of chemotherapy or diabetes.”
Multiple underlying causes
They emphasised determining the cause of chronic cough is crucial to effective treatment. In many cases, more than one underlying condition may cause a chronic cough.
Healthcare professionals pointed out a persistent cough sometimes goes beyond being a mere inconvenience, as it can disrupt one’s sleep and lead to feelings of exhaustion. In more severe instances, chronic coughing may induce vomiting and dizziness, and even rarely result in rib fractures.
Dr Zaid Mahdi Mohammed, Canadian Specialist Hospital Dubai, said, “The most common causes of chronic cough are postnasal drip, asthma, and acid reflux from the stomach. These three causes are responsible for up to 90 per cent of all cases of chronic cough. Honey and saltwater gargling, using a humidifier, or taking steam can be some of the effective home remedies.”
Respiratory Devices And Equipment (Therapeutic) Market
The Business Research Company has updated its global market reports, featuring the latest data for 2024 and projections up to 2033
The Business Research Company offers in-depth market insights through Respiratory Devices And Equipment (Therapeutic) Global Market Report 2024, providing businesses with a competitive advantage by thoroughly analyzing the market structure, including estimates for numerous segments and sub-segments.
Market Size And Growth Forecast:
The respiratory devices and equipment (therapeutic) market size has grown rapidly in recent years. It will grow from $19.9 billion in 2023 to $22.01 billion in 2024 at a compound annual growth rate (CAGR) of 10.6%. The growth in the historic period can be attributed to respiratory conditions prevalence, aging population, technological advancements, respiratory disease management.
The respiratory devices and equipment (therapeutic) market size is expected to see rapid growth in the next few years. It will grow to $32.61 billion in 2028 at a compound annual growth rate (CAGR) of 10.3%. The growth in the forecast period can be attributed to chronic diseases and lifestyle factors, telehealth expansion, technological innovations, copd management. Major trends in the forecast period include digital health and remote monitoring, ventilators and life support systems, respiratory rehabilitation, telehealth integration.
The main products of the respiratory devices and equipment (therapeutic) market are nebulizers, humidifiers, oxygen concentrators, positive airway pressure devices, ventilators, capnographs, and gas analyzers. A nebulizer is a small machine that turns liquid medicine into a mist, sits with the machine, and breathes in by a connected mouthpiece. The various technologies involved in the respiratory devices and equipment are HEPA filter, electrostatic filtration, microsphere separation, hollow fiber filtration, and others. The market covered in this report is segmented by end-users into home care settings and hospitals.
Major Driver - Rising Respiratory Disease Prevalence Drives Respiratory Devices And Equipment (Therapeutic) Market
The rising prevalence of respiratory diseases such as chronic obstructive pulmonary disorder (COPD) and sleep apnea contributed to the growth of the therapeutic respiratory devices and equipment market. According to World Health Organization, one million people die due to chronic obstructive pulmonary diseases caused by smoking among the 4.9 million people who die due to tobacco consumption and 65 million people suffer from moderate to severe COPD. As per its estimates, COPD is predicted to be the third leading cause of death worldwide and potentially fatal respiratory diseases. Tuberculosis, COPD, and lung cancer will account for about one in five deaths worldwide by 2030. According to National Health Interview Survey by the Centers for Disease Control and Prevention (CDC), the number of adults with diagnosed chronic bronchitis in the USA was 9.0 million. In the USA, it is estimated that 22 million Americans suffer from sleep apnea, with 80% of the cases of moderate and severe obstructive sleep apnea undiagnosed. According to a research study published on American Journal of Respiratory and Critical Care Medicine, in 2021, global prevalence of obstructive sleep apnea was found to be around 20% globally. The increased prevalence of COPD and sleep apnea in the geriatric population is driving the growth of the respiratory devices and equipment (therapeutic) market.
Competitive Landscape:
Major companies operating in the respiratory devices and equipment (therapeutic) market include Hamilton Medical AG, Koninklijke Philips N.V., Smiths Medical, GE Healthcare, Philips Healthcare, Chart Industries Inc., Invacare Corporation, Fisher & Paykel Healthcare Limited, ResMed Inc., Drägerwerk AG & Co. KGaA, Medtronic plc, Masimo Corporation, CareFusion Corporation, Getinge AB, Hill-Rom Holdings Inc., Becton, Dickinson and Company, Air Liquide S.A., Vyaire Medical Inc., Compumedics Limited, Covidien plc, Mindray Medical International Limited, Inogen Inc., Nihon Kohden Corporation, Siemens Healthcare GmbH, Teleflex Incorporated, Breas Medical AB, AirSep Corporation, Rotech Healthcare Inc., 3B Medical Inc.
Top Trend - Innovations In Artificial Intelligence (AI)-Enhanced Respiratory Devices And Monitoring
The companies in the respiratory devices and equipment (therapeutic) market are increasingly using AI to develop respiratory devices to treat Asthma and COPD. Artificial intelligence supports the development of innovative sensors-equipped inhalers which help patients to track their dosage intake. These sensors are durable and consume less power and help caution the patients by noting the differences or fluctuations in respiration. These are used for both add-on and embedded inhalers. These inhalers with sensors can track data automatically and can alert both the doctors and patients about the health condition of the patients. Also, the companies in developing devices such as AI aided imaging systems and AI aided platforms that will act as voice biomarkers. For instance, in March 2022, Respira Labs, a US-based respiratory technology company, launched AI-powered wearable lung monitor 'Sylvee'. The new product uses acoustic resonance to assess the lung function and detect any variations in the lung function.
The Table Of Content For The Market Report Include:
1. Executive Summary
2. Respiratory Devices And Equipment (Therapeutic) Market Characteristics
3. Respiratory Devices And Equipment (Therapeutic) Market Trends And Strategies
The Business Research Company (www.thebusinessresearchcompany.com) is a market intelligence firm that pioneers in company, market, and consumer research. Located globally, TBRC's consultants specialize in various industries including manufacturing, healthcare, financial services, chemicals, and technology. The firm has offices located in the UK, the US, and India, along with a network of proficient researchers in 28 countries.
Pierachille Santus,1 Fabiano Di Marco,2 Fulvio Braido,3 Marco Contoli,4 Angelo Guido Corsico,5 Claudio Micheletto,6 Girolamo Pelaia,7 Dejan Radovanovic,1 Paola Rogliani,8 Laura Saderi,9 Nicola Scichilone,10 Silvia Tanzi,11 Manlio Vella,11 Silvia Boarino,11 Giovanni Sotgiu,9 Paolo Solidoro12
1Department of Biomedical and Clinical Sciences (DIBIC), Università degli Studi di Milano, Division of Respiratory Diseases, Ospedale L. Sacco, ASST Fatebenefratelli-Sacco, Milano, Italy; 2Department of Health Sciences, Università degli Studi di Milano Pneumology, ASST Papa Giovanni XXIII, Bergamo, Italy; 3Department of Internal Medicine (DiMI), Respiratory Unit for Continuity of Care, IRCCS Ospedale Policlinico San Martino, University of Genova, Genova, Italy; 4Department of Translational Medicine, Respiratory Section, University of Ferrara, Ferrara, Italy; 5Department of Medical Sciences and Infective Diseases, Unit of Respiratory Diseases, IRCCS Policlinico San Matteo Foundation and University of Pavia Medical School, Pavia, Italy; 6Cardio-Thoracic Department, Respiratory Unit, University Integrated Hospital, Verona, Italy; 7Dipartimento di Scienze della Salute, Università Magna Graecia, Catanzaro, Italy; 8Department of Experimental Medicine, Unit of Respiratory Medicine, University of Rome ”Tor Vergata”, Division of Respiratory Medicine, University Hospital ”Tor Vergata”, Rome, Italy; 9Department of Medicine, Surgery and Pharmacy, University of Sassari, Sassari, Italy; 10Biomedical Department of Internal and Specialist Medicine, University of Palermo, Palermo, Italy; 11AstraZeneca Italia, Milan, Italy; 12Department of Medical Sciences, University of Turin, S.C. Pneumologia, Azienda Ospedaliero Universitaria Città della Salute e della Scienza, Torino, Italy
Correspondence: Pierachille Santus, Università degli Studi di Milano, Via G.B. Grassi 74, Milano, 20157, Italy, Tel +39 0239042801, Fax +39 0239042473, Email [email protected]
Objective: To describe the burden of moderate to severe exacerbations and all-cause mortality; the secondary objectives were to analyze treatment patterns and changes over follow-up. Design: Observational, multicenter, retrospective, cohort study with a three year follow-up period. Setting: Ten Italian academic secondary- and tertiary-care centers. Participants: Patients with a confirmed diagnosis of COPD referring to the outpatient clinics of the participating centers were retrospectively recruited. Primary and Secondary Outcome Measures: Annualized frequency of moderate and severe exacerbations stratified by exacerbation history prior to study enrollment. Patients were classified according to airflow obstruction, GOLD risk categories, and divided in 4 groups: A = no exacerbations; B = 1 moderate exacerbation; C = 1 severe exacerbation; D = ≥ 2 moderate and/or severe exacerbations. Overall all-cause mortality stratified by age, COPD category, and COPD therapy. A logistic regression model assessed the association of clinical characteristics with mortality. Results: 1111 patients were included (73% males), of which 41.5% had a history of exacerbations. As expected, the proportion of patients experiencing ≥ 1 exacerbation during follow-up increased according to pre-defined study risk categories (B: 79%, C: 84%, D: 97.4%). Overall, by the end of follow-up, 45.5% of patients without a history of exacerbation experienced an exacerbation (31% of which severe), and 13% died. Deceased patients were significantly older, more obstructed and hyperinflated, and more frequently active smokers compared with survivors. Severe exacerbations were more frequent in patients that died (23.5%, vs 10.2%; p-value: 0.002). Chronic heart failure and ischemic heart disease were the only comorbidities associated with a higher odds ratio (OR) for death (OR: 2.2, p-value: 0.001; and OR: 1.9, p-value: 0.007). Treatment patterns were similar in patients that died and survivors. Conclusion: Patients with a low exacerbation risk are exposed to a significant future risk of moderate/severe exacerbations. Real life data confirm the strong association between mortality and cardiovascular comorbidities in COPD.
Keywords: pulmonary disease chronic obstructive, heart failure, ischaemic heart disease, respiratory medicine, public health
Introduction
Chronic obstructive pulmonary disease (COPD) is a treatable but debilitating medical condition associated with persistent symptoms and chronic airflow obstruction.1 Despite the availability of multiple therapeutic options, COPD is the third leading cause of death worldwide and has a substantial socioeconomic impact.2,3 COPD is diagnosed when patients present with respiratory symptoms and/or history of exposure to risk factors, having bronchial obstruction confirmed by spirometry.2 However, even mild obstruction hides a significant loss of small airways4 making a timely diagnosis and a prompt treatment initiation of great importance to reduce morbidity and mortality.5 Greater understanding of individual variability of COPD progression through multidimensional evaluation may help recommend tailored interventions.6–9 Patients with COPD are susceptible to exacerbations, in fact, 30%-50% of patients experience at least one exacerbation per year.9 Exacerbations are associated with disease severity and history of previous exacerbations itself is considered the most reliable predictor of future exacerbations.10 Nevertheless, patients with mild airflow obstruction and symptoms that may not yet affect activities of daily living can still experience frequent or severe exacerbations.11 Also, mild and moderate exacerbations can increase the risk of future exacerbations, accelerating lung function decline, promoting cardiovascular complications, and increasing mortality.12–14
A Canadian study showed that severe exacerbations leading to hospitalization may increase the risk of a second severe event by 3-fold and may increase mortality up to 50% after 3.6 years of follow-up after a first hospitalization.15 Moreover, after a severe exacerbation, patients are at greater risk of cardiovascular events,16,17 putting pharmacological and non-pharmacological preventive strategies the highest priority in the management of the disease.18 Pharmacological options include long-acting β2 agonists (LABA) and/or long-acting antimuscarinic agents (LAMA), in combination or without inhaled corticosteroids (ICS), that decrease airway inflammation and reduce the rate of exacerbations.19,20
The estimated prevalence of COPD in Italy ranges from 2.6%, assessed via patient-directed survey,21 to 3.01% in primary care,22 thus affecting up to 3.5 million adults and representing the sixth most prevalent chronic disease. It also has large impact on the national healthcare system: the mean annual cost per patient was €3291 in 2015, with the major cost component being hospitalizations following exacerbations.23 Considering the overall socio-economic and healthcare burden of the disease, a detailed clinical profile of COPD patients in Italy appears desirable, but unfortunately to date, real life data are lacking. The present real life study is aimed at describing the clinical and functional characteristics, treatment patterns, impact of exacerbations and comorbidities and their association with mortality in a large cohort of Italian patients with COPD.
Materials and Methods
Study Design
The DescribinG bUrden of COPD and occurrence of mortaLity in a cohort of Italian Patients (GULP) study, part of AstraZeneca’s European AvoidEX program, was an observational, multicenter, retrospective cohort study based on a multicenter database, recently approved as the Italian COPD Registry (Ethics Committee protocol n. 20–27 Sept 2023), conducted in ten Italian academic secondary- and tertiary-care centers: Division of Respiratory Diseases of L. Sacco University Hospital (Università degli Studi di Milano, Milano), Pneumology unit, ASST Papa Giovanni XXIII (Università degli Studi di Milano, Bergamo), Respiratory Unit for Continuity of Care, IRCCS Ospedale Policlinico San Martino (University of Genova, Genova), Respiratory Section, Department of Translational Medicine (University of Ferrara, Ferrara), Unit of Respiratory Diseases, IRCCS Policlinico San Matteo Foundation (University of Pavia Medical School, Pavia), Respiratory Unit, Cardio-Thoracic Department (University Integrated Hospital, Verona), Pulmonary Unit, Dipartimento di Scienze della Salute (Università Magna Graecia, Catanzaro), Unit of Respiratory Medicine, Department of Experimental Medicine (University of Rome “Tor Vergata”, Rome), Biomedical Department of Internal and Specialist Medicine (University of Palermo, Palermo), Pulmonary Unit, Azienda Ospedaliero Universitaria Città della Salute e della Scienza (University of Turin, Torino). The study was carried out according to the amended Declaration of Helsinki, ICH GCPs, GPP, and the legislation on non-interventional studies and/or observational studies (AIFA guidelines, 20/Mar/2008) and approved by the ethics committee of each participating site. All participants gave written informed consent.
Data protection and privacy legislation compliance were ensured. The dataset covered a period of 365 days prior to the index date and a minimum of 365 days post index date up to three years of follow up. The index date was the date of the study entry, i.e. the date when the patient entered the database with a record of a COPD diagnosis.
Study Objectives
The primary objective was to describe the burden of moderate to severe COPD exacerbations. Rates of moderate and severe exacerbations, as well as all-cause mortality were collected and analyzed.
The secondary objective was to describe treatment patterns at baseline and eventual treatment changes.
The pharmacological inhaled treatments considered were: LAMA or LABA monotherapies or fixed combinations thereof, ICS and a LABA and/or a LAMA or their fixed combinations.
Mortality was assessed at 3 years. Patients were stratified in two groups according to survival status and the following variables were assessed: demographic and clinical characteristics, baseline exacerbations, relationship between mortality and clinical characteristics.
Study Subjects
Electronic records of patients aged ≥40 years with an established diagnosis of COPD between January 1, 2015, and December 31, 2017, and referring to the outpatient clinics of the participating centers were retrospectively reviewed. COPD diagnosis was considered if having age ≥ 40 years old, a smoking history > 20 pack years and a post-bronchodilator forced expiratory volume in one second to slow vital capacity ratio (FEV1/VC) < the lower limit of normal (LLN) criteria.11 Severity of disease was graded using three different classifications proposed by GOLD over time: airflow obstruction (GOLD stages 1 to 4);24 airflow obstruction, exacerbations and respiratory symptoms25 or exacerbations and respiratory symptoms (GOLD A, B, C, or D).26 Patients were excluded if had a current asthma diagnosis or clinically significant alternative respiratory diseases such as interstitial lung disease or bronchiectasis.
Clinical phenotypes of the enrolled patients were obtained following the multifactorial model proposed by Pistolesi et al.27 The presence of chronic cough, sputum, and sputum purulence, adventitious sounds and hyper-resonance at physical examination, chest X-ray parameters, such as increased vascular markings, bronchial wall thickening, increased lung volume and reduced lung density, together with the FEV1/FVC ratio were registered. These parameters were included in the web-based estimation model28 that allowed the assessment of the predominant clinical phenotype: airways obstructive (chronic bronchitis), parenchymal destructive (emphysema), or intermediate. At enrollment, patients were assigned to one of 6 groups based on the ongoing therapy:
LAMA or LABA monotherapy
Combinations of LABA + LAMA
ICS without LABA or LAMA
Combinations of ICS + LABA or ICS + LAMA
Combinations ICS + LABA + LAMA
None of the above
Patients treated with more than one pharmacological class were considered as exposed to combination therapy if they had taken the medications for at least 14 days prior to the index date.
Outcomes and Variables
According to the history of exacerbations in the year before the index date,29 patients were grouped into one of four categories:
Category A: no exacerbations
Category B: 1 moderate exacerbation (symptomatic deterioration requiring antibiotic therapy or Medium to high-dose systemic corticosteroids)
Category C: 1 severe exacerbation (exacerbation requiring hospitalization or emergency visits)
Category D: ≥2 moderate and/or severe exacerbations
Moderate exacerbations were defined as claims for courses of oral corticosteroids and/or respiratory antibiotics. Severe exacerbations were defined as need for hospitalization. If more than one of the episodes occurred within a 2-week window, a single exacerbation was considered. If a moderate and a severe exacerbations occurred concurrently within a 2-week window, the episode was considered as a severe exacerbation.
Patient and Public Involvement
Due to the study design, patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of our research.
Statistical Analysis
Categorical variables were summarized with absolute and relative frequencies. Continuous variables were summarized with central tendency (i.e. medians) and variability (i.e. interquartile ranges, IQR) indicators. Statistical differences were evaluated using chi-square or Mann–Whitney tests, as appropriate. COPD exacerbations are described overall and in selected strata. All-cause mortality is described overall and stratified by age (<65, 65–75, >75), COPD category, and COPD therapy. A logistic regression model was used to evaluate association of covariates at enrollment with mortality. Missing data were not imputed. All statistical analyses were performed using the statistical software STATA version 16 (StatsCorp, Texas, USA).
Results
Characteristics of the Study Population
The study included 1111 COPD patients (Table 1). Patients were predominately male (72.9%) with a median (IQR) age of 76 (70–82) years and body mass index of 26.5 (23.4–29.4) Kg/m2. Most participants were current smokers (70.1%) with a median (IQR) smoking history of 40 (30–60) pack-years. 56.8% of patients had emphysema and 14.9% had chronic bronchitis, whereas 28.4% had a mixed phenotype. Most patients had moderate to severe airflow obstruction (GOLD 2 and GOLD 3: 44.8% and 28.1%, respectively). By GOLD 2016 and GOLD 2017 criteria, the highest proportion of patients was classified as GOLD D (46.4% and 41.4%, respectively) (Table 2). Among the COPD therapies, the most widely prescribed were LABA+LAMA (21% in 2015; 25.9% in 2016, and 28.7% in 2017) and combination therapy with ICS +LABA+LAMA (42.4% in 2015; 44.5% in 2016, and 46.6% in 2017). 13.3% of patients died within three years of follow-up.
Table 1 Patients’ Characteristics at Enrollment
Table 2 Exacerbations and Treatment Patterns During the Follow-Up
Exacerbation Patterns in COPD Patients
Prior to the index date, 41.5% (461/1111) of patients had a history of exacerbations. During follow up the majority of patients experienced moderate exacerbations (37.3%, 41.4%, and 40% for each year of follow up, respectively) (Table 2), while the proportion of patients experiencing severe exacerbations was lower though stable over the follow up period (17.8%, 18.9%, 15.3%).
Among patients without prior history of exacerbations (category A), 45.5% experienced an exacerbation during the follow up, 30.6% of which were severe. The proportion of patients with an exacerbation during the follow up period increased in categories B, C and D (60.7%, 83.7%, 97.4%, respectively). Accordingly, the proportion of moderate exacerbations during follow up increased with increasing exacerbation risk from category A to D (28.4%, 60.7%, 58.2%, 90.6%, respectively). Patients that were frequent exacerbators in the year before entering the study (group D) experienced the highest median number of moderate exacerbations during follow up (4 (2–5); p<0.001 compared with other groups). 18.5% of patients with a history of one moderate exacerbation in the previous year (category B) had a severe exacerbation during the follow up. The highest proportion of severe exacerbations was observed in patients with a single severe (category C, 51%) and frequent exacerbators (category D, 34.6%) (Table 3).
Table 3 Exacerbations Over 3 Years in a, B, C and D Categories
COPD Treatments and Therapeutic Switch
A significant percentage of patients switched inhaled therapy by the end of the follow up period (Table 4). Patients on a bronchodilator monotherapy most frequently switched to a LABA/LAMA combination (38.7% of patients previously on a LABA and 22% of patients on a LAMA) (Table 4). The proportion of patients already on LABA/LAMA and on LABA/ICS that switched to a triple combination therapy (ICS/LABA/LAMA) was 14.8% and 28.2% respectively, while 81.8% of patients treated with LABA/LAMA continued the same therapy, a proportion that increased to 89% in patients treated with ICS/LABA/LAMA. ICS were withdrawn in 21.8% of cases in patients treated with ICS/LABA, while this percentage was reduced to 11.1% in patients on ICS/LABA/LAMA, the majority of which (8.7%) were switched to a LABA/LAMA combination (Table 4).
Table 4 Pharmacological Therapy for COPD, 2015 Vs 2017
Characteristics of Deceased Patients
Compared to patients alive at the end of the follow up, patients who died were significantly older, more frequently active smokers, and were significantly more obstructed and hyperinflated (Supplementary Table 1). The proportion of patients that experienced at least one exacerbation during follow up did not differ between groups, but the proportion of patients experiencing moderate exacerbations tended to be less (13.7% vs 16.8%) while severe exacerbations were significantly more frequent in patients that did not survive (23.5%, vs 10.2%; p-value: 0.002) (Supplementary Table 1). Frequent exacerbators were similar between groups (20.6% vs 23.2%). The distribution of treatment patterns at the end of follow up was not different in patients that died and those that survived, although the proportion of patients on ICS/LABA/LAMA tended to be higher in the former group (59.7% vs 47.9%). Cardiovascular comorbidities were the most frequently observed, being significantly more prevalent in deceased patients than in patients alive at the end of follow up (27.2% vs 14.2% for chronic heart failure, p-value: <0.0001; 28.1% vs 17.3% for ischemic heart disease, p-value: 0.006) (Supplementary Table 1). Chronic heart failure and ischemic heart disease were the only comorbidities/clinical characteristics associated with a significantly higher odds ratio (OR) for death (OR: 2.2, p-value: 0.001; and OR: 1.9, p-value: 0.007, respectively) (Figure 1). Mortality was significantly higher in patients with a history of one severe exacerbation (category C): 24.7% VS category A (10.7%), category B (10.4%) and category D (11.2%) (Supplementary Figure 1).
Figure 1 Association between baseline descriptors and mortality outcome during follow-up: multivariable logistic regression model. The forest plot illustrates the odds of mortality with 95% confidence intervals (CI). CI and p-values are reported on the left of y axis. Values higher than 1 favor risk of death.
Abbreviations: BMI, body mass index; IC, inspiratory capacity; IQR, inter quartile range; FEV1, forced expiratory volume in first second.
Discussion
The present study evaluated clinical characteristics, treatment patterns, rates of moderate and severe exacerbations, and survival of a cohort of Italian COPD patients.
The burden of exacerbations was almost constant during the study period: 45.5% of patients that had no exacerbations in the year before entering the study experienced at least one exacerbation over a 3-year follow-up period. Moreover, 79.3% of patients that already had a history of a moderate exacerbation had at least one subsequent event. This suggests that even patients perceived as low-risk should be adequately managed over time, since the absence of previous events in the majority of cases does not prevent the occurrence of future exacerbations, highlighting the importance of preventing exacerbation of any severity in order to reduce the risk of future events, and monitoring progression and preventing worsening of disease represent crucial goals, considering that moderate exacerbations correlate with a high risk of severe exacerbations and increased mortality.15 Indeed, in the present study, mortality was associated with severe exacerbations, which confirms the importance of exacerbation events in prognosis.
COPD mostly affects older adults, and the development of multimorbidity may complicate COPD management.30 People living with COPD have almost twice the risk of heart failure and myocardial infarction when compared with those without COPD belonging to the same age, sex, race, and education level.30 Even COPD patients with no history of cardiovascular disease have a higher risk of cardiovascular complications, such as myocardial infarction and stroke, following a moderate exacerbation.14 We showed that cardiovascular comorbidities are a major risk factor for death in Italian patients with COPD. In fact, among all comorbidities, only chronic heart failure and ischemic heart disease were associated with a significantly higher risk of death, independent of the severity of airflow obstruction or hyperinflation. Apparently, frequent exacerbators (group D) were exposed to a lower risk of hospitalizations compared with group C during follow up. Considered the higher mortality in group C and the proportion of patients with cardiovascular comorbidities among patients that died, it could be speculated that patients with frequent exacerbations, irrespective of the severity of exacerbations, could be exposed to a stricter pulmonary outpatient monitoring and therefore with a higher chance of being managed outside the hospital setting in case of an exacerbation. On the other hand, patients in group C might have had a higher risk of being hospitalized for an acute event, with an increased overall mortality risk secondary to the higher prevalence of cardiovascular risk factors.
Current treatments for COPD foresee escalation of therapy from monotherapy to dual/triple therapy based on symptoms and number and severity of exacerbations, and is usually recommended in symptomatic patients with a history of frequent and/or severe exacerbations.2 ICS/LABA/LAMA fixed-dose combinations improve respiratory function, symptoms, health status, and reduce exacerbations compared to dual therapies.31,32 Triple therapy also demonstrated a significant impact on mortality and frequency of moderate or severe exacerbations compared with LABA/LAMA.2,33,34 Our observations showed that patients treated with triple therapy remained on triple therapy throughout the study, whereas patient prescribed ICS/LABA were often stepped up to triple therapy. In spite of the recommendation for ICS treatment only in patients that experience exacerbations,2 in our real life study we observed that the prevalence of ICS prescription in clinical practice reaches 50% of the patients enrolled, suggesting the possibility of overtreatment or inadequate disease control despite maximized bronchodilation in a proportion of patients.
Our work demonstrated a high prevalence of cardiovascular comorbidities in patients with COPD, confirming previous observations.35,36 Furthermore, our analysis showed that after three years of follow up a notable percentage of patients died (13.3%) and only chronic heart failure and ischemic heart disease were associated with higher odds of mortality. Patients that died during the follow up had poorer lung function (lower FEV1 and inspiratory capacity) and had more frequently a history of a severe exacerbation before entering the study, thus justifying the higher proportion treated with triple therapy, but also suggesting that triple therapy is initiated late in the clinical history pf COPD patients. These observations confirm the need for increased alertness on pharmacological optimization and careful patients’ assessment in terms of exacerbations and mortality, and on the connection between chronic cardiac and lung diseases, in order to improve both patients’ quality and quantity of life.
The present study has several limitations. First, patients were enrolled from secondary and tertiary care hospitals, thus the study might suffer from a selection bias, making results not fully generalizable in terms of severity of disease and mortality. Second, in the last years prescription patterns have changed over time due to the market introduction of triple fixed dose combination therapies, therefore switching patterns might have evolved differently than described. Third, the cause of death was not registered therefore any consideration about the possible causative role of cardiovascular comorbidities or exacerbations in the risk of death could not be drawn. Finally, adverse drug effects and major cardiovascular events were not studied and the cause of therapeutic switch was not assessed. Indeed, the study has strengths, mainly represented by the real life setting, the multicenter study and by the length of the follow up period.
Conclusion
In conclusion, this study provided for the first time a detailed clinical overview of the exacerbation burden in patients with COPD in Italy, highlighting from real life data that even patients with a low exacerbation risk are exposed to a significant future risk of moderate to severe exacerbations. The study also confirmed the existence of a strong association between mortality and cardiovascular comorbidities in COPD, in particular with heart failure and ischemic heart disease. Despite the overall exacerbation and mortality burden, a lower than expected number of patients were treated with triple therapy with ICS//LABA/LAMA. The study should represent a starting point and gives the rationale for continuing the implementation of large shared national databases as a source of patients’ characterization and as monitoring tools for preventive pharmacological and non-pharmacological strategies.
Data Sharing Statement
The anonymized dataset will be available upon reasonable request by the Corresponding Author.
Acknowledgments
Medical writing and editorial assistance were provided by Maria Vittoria Verga Falzacappa, PhD (EDRA S.p.A., Milan, Italy) and funded by AstraZeneca.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Astra Zeneca.
Disclosure
PSa has received lectures fees at national and international meetings and consultancy fees from Boehringer Ingelgheim, Chiesi Farmaceutici, Astra Zeneca, Berlin-Chemie, Edmondpharma, Guidotti, Neopharmed, Novartis, Valeas, GlaxoSmithKline, Alfasigma, Zambon and Sanofi; research grants from Air Liquide, Almirall, Boehringer Ingelgheim, Chiesi Farmaceutici, Pfizer, Edmondpharma. FB declares participation in a company sponsored speaker’s bureau: Astra Zeneca, GSK, Novartis, Boehringer Ingelgheim, Chiesi, MSD, Menarini, Malesci, Guidotti, Sanofi and support for research: Chiesi, Vitalair. M.C. declares grants for research, personal fees and non-financial support from Chiesi and GlaxoSmithKline, personal fees and non-financial support from AstraZeneca, Boehringer Ingelheim, Alk-Abello, and Novartis, and research grants from the University of Ferrara, Italy. FDM has received lectures fees at national and international meetings and consultancy fees from Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi Farmaceutici, Dompe, Guidotti/Malesci, GlaxoSmithKline, Menarini, Novartis, and Zambon; CM received fees as a speaker from Astrazeneca, GSK, Sanofi, Chiesi, Menarini, Guidotti, Novartis, Zambon, Boehringer. GP has received lecture fees and consultancy fees from Alfasigma, AstraZeneca, Chiesi, GlaxoSmithKline, Guidotti-Malesci, Menarini, Mundipharma, Novartis, Sanofi, Zambon. DR has received fees for lectures from Astra Zeneca, Berlin Chemie, Boehringer Ingelheim, Glaxo Smith Kline, Menarini; fees for consultancy from Damor Farmaceutic and honoraria for consulting and participation to advisory boards from Astra Zeneca, Boehringer Ingelheim. PR participated as a lecturer and advisor in scientific meetings and courses under the sponsorship of Almirall, AstraZeneca, Biofutura, Boehringer Ingelheim, Chiesi Farmaceutici, GlaxoSmithKline, Menarini Group, MSD, Mundipharma, Novartis and Recipharm. Her department was funded by Almirall, Boehringer Ingelheim, Chiesi Farmaceutici, Novartis, and Zambon. NS has received lectures fees at national and international meetings and consultancy fees from Astra Zeneca, Boehringer Ingelheim, Chiesi Farmaceutici, GlaxoSmithKline; research grants from Boehringer Ingelheim, Chiesi Farmaceutici, Sanofi. SB, ST, MV are AstraZeneca employees. PSo has participated as a lecturer, speaker, and advisor in scientific meetings and courses under the sponsorship from Boehringer Ingelheim, Chiesi Farmaceutici, Astra Zeneca, Guidotti-Malesci, Novartis, Valeas, GlaxoSmithKline, Menarini, ABC Farmaceutici, Almirall, Dompè and Biotest. The authors report no other conflicts of interest in this work.
References
1. Viegi G, Pistelli F, Sherrill DL, et al. Definition, epidemiology and natural history of COPD. Eur Respir J. 2007;30(5):993–1013. doi:10.1183/09031936.00082507
2. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. 2022.Available from: goldcopd.org/2022-gold-reports-2/. Accessed October12, 2022.
3. Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012;380(9859):2095–2128. doi:10.1016/S0140-6736(12)61728-0
4. McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–1575. doi:10.1056/NEJMoa1106955
5. Radovanovic D, Contoli M, Braido F, et al. Future perspectives of revaluating mild COPD. Respiration. 2022;101(7):688–696. doi:10.1159/000524102
6. Papaioannou AI, Loukides S, Gourgoulianis KI, et al. Global assessment of the COPD patient: time to look beyond FEV1? Respir Med. 2009;103(5):650–660. doi:10.1016/j.rmed.2009.01.001
7. Casanova C, de Torres JP, Aguirre-Jaime A, et al. The progression of chronic obstructive pulmonary disease is heterogeneous: the experience of the BODE cohort. Am J Respir Crit Care Med. 2011;184(9):1015–1021. doi:10.1164/rccm.201105-0831OC
8. Han MK, Wise R, Mumford J, et al. Prevalence and clinical correlates of bronchoreversibility in severe emphysema. Eur Respir J. 2010;35(5):1048–1056. doi:10.1183/09031936.00052509
9. Whittaker H, Rubino A, Mullerova H, et al. Frequency and severity of exacerbations of COPD associated with future risk of exacerbations and mortality: a UK routine health care data study. Int J Chron Obstruct Pulmon Dis. 2022;17:427–437. doi:10.2147/COPD.S346591
10. Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363(12):1128–1138. doi:10.1056/NEJMoa0909883
11. Radovanovic D, Contoli M, Di Marco F, et al. Clinical and functional characteristics of COPD patients across gold classifications: results of a multicenter observational study. COPD. 2019;16(3–4):215–226. doi:10.1080/15412555.2019.1659760
12. Alqahtani JS, Aquilina J, Bafadhel M, et al. Research priorities for exacerbations of COPD. Lancet Respir Med. 2021;9(8):824–826. doi:10.1016/S2213-2600(21)00227-7
13. Celli BR, Decramer M, Wedzicha JA, et al. An official American thoracic society/European respiratory society statement: research questions in COPD. Eur Respir J. 2015;45(4):879–905. doi:10.1183/09031936.00009015
14. Donaldson GC, Hurst JR, Smith CJ, et al. Increased risk of myocardial infarction and stroke following exacerbation of COPD. Chest. 2010;137(5):1091–1097. doi:10.1378/chest.09-2029
15. Suissa S, Dell’Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax. 2012;67(11):957–963. doi:10.1136/thoraxjnl-2011-201518
16. Hesse K, Bourke S, Steer J. Heart failure in patients with COPD exacerbations: looking below the tip of the iceberg. Respir Med. 2022;196:106800. doi:10.1016/j.rmed.2022.106800
17. Dransfield MT, Criner GJ, Halpin DMG, et al. Time-dependent risk of cardiovascular events following an exacerbation in patients with chronic obstructive pulmonary disease: post hoc analysis from the IMPACT trial. J Am Heart Assoc. 2022;11(18):e024350. doi:10.1161/JAHA.121.024350
18. Halpin DM, Miravitlles M, Metzdorf N, et al. Impact and prevention of severe exacerbations of COPD: a review of the evidence. Int J Chron Obstruct Pulmon Dis. 2017;12:2891–2908. doi:10.2147/COPD.S139470
19. Burge PS, Calverley PM, Jones PW, et al. Randomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: the ISOLDE trial. BMJ. 2000;320(7245):1297–1303. doi:10.1136/bmj.320.7245.1297
21. Ferrante G, Baldissera S, Campostrini S. Epidemiology of chronic respiratory diseases and associated factors in the adult Italian population. Eur J Public Health. 2017;27(6):1110–1116. doi:10.1093/eurpub/ckx109
22. Lupi L. Prevalenza della broncopneumopatia cronica ostruttiva e pattern di utilizzo vaccino antinfluenzale nei pazienti assistititi dalla Medicina Generale Italiana. Newsletter Health Search Istituto di Ricerca della S.I.M.G. (Società Italiana di Medicina Generale e delle Cure Primarie), 2020. Available from: www.healthsearch.it/documenti/documenti_ricercatori/Newsletter/2020/1_2020.pdf. Accessed October20, 2022.
23. Dal negro RW. COPD: the annual cost-of-illness during the last two decades in Italy, and its mortality predictivity power. Healthcare. 2019;7(1):35. doi:10.3390/healthcare7010035
27. Pistolesi M, Camiciottoli G, Paoletti M, et al. Identification of a predominant COPD phenotype in clinical practice. Respir Med. 2008;102(3):367–376. doi:10.1016/j.rmed.2007.10.019
28. Clinical Identification of Phenotypes in COPD. CLIP COPD 2022; 2022. www.clipcopd.com/. Accessed October20, 2022.
30. Witt LJ, Wroblewski KE, Pinto JM, et al. Beyond the lung: geriatric conditions afflict community-dwelling older adults with self-reported chronic obstructive pulmonary disease. Front Med Lausanne. 2022;9:814606. doi:10.3389/fmed.2022.814606
31. Solidoro P, Albera C, Ribolla F, et al. Triple therapy in COPD: can we welcome the reduction in cardiovascular risk and mortality? Front Med Lausanne. 2022;9:816843. doi:10.3389/fmed.2022.816843
32. Calzetta L, Cazzola M, Matera MG, et al. Adding a LAMA to ICS/LABA therapy: a meta-analysis of triple combination therapy in COPD. Chest. 2019;155(4):758–770. doi:10.1016/j.chest.2018.12.016
33. Lai CC, Chen CH, Chen KH, et al. The impact of 52-week single inhaler device triple therapy versus dual therapy on the mortality of COPD patients: a systematic review and meta-analysis of randomized controlled trials. Life. 2022;12(2):173. doi:10.3390/life12020173
34. Rabe KF, Martinez FJ, Ferguson GT, et al. Triple inhaled therapy at two glucocorticoid doses in moderate-to-very-severe COPD. N Engl J Med. 2020;383(1):35–48. doi:10.1056/NEJMoa1916046
35. Rogliani P, Ritondo BL, Laitano R, Chetta A, Calzetta L. Advances in understanding of mechanisms related to increased cardiovascular risk in COPD. Expert Rev Respir Med. 2021;15(1):59–70. doi:10.1080/17476348.2021.1840982
36. Decramer M, Janssens W. Chronic obstructive pulmonary disease and comorbidities. Lancet Respir Med. 2013;1(1):73–83. doi:10.1016/S2213-2600(12)70060-7
Filtering face-piece respirators like N95 masks are often a standard piece of personal protective equipment (PPE) in workplaces where people are exposed to dust and other airborne hazards. But PPE isn’t just for the workplace. Home maintenance and DIY work can also expose you to particles in the air that can harm your health.
“Whether you’re a trained construction worker or a DIY enthusiast, you could be putting yourself at risk for serious respiratory diseases if you don’t use appropriate protection,” states Dr. Chaitanya Mandapakala, Medical Director of Chronic Lung Diseases at St. Elizabeth Healthcare.
Long-term exposure to airborne particles has been linked to respiratory diseases, infections, and cancer. While exposure to airborne hazards during home projects is generally much lower than that of a worker who spends every day in dust-filled environments, DIYers also need to protect their respiratory systems by wearing an appropriate respirator for the work they’re doing.
What Are N95 Masks?
N95 masks are air-purifying respirators designed to filter out 95% of particulates with a diameter of 0.3μm or larger. “Particulates are basically any small particles present in the air naturally, such as pollen, mold, bacteria, or wind-blown dust, or those generated during work processes, such as grinding, sanding, cutting, welding, crushing, cutting, sweeping, dusting, spraying, or burning,” explains Curtis Chambers, Board-Certified Safety Professional (CSP) and president of OSHA Training Services Inc.
If you still have a stack of N-95s at home, they can go to good use by protecting you from harmful particulates during DIY work. “A clean, well-fitting N95 respirator will generally offer adequate protection against most airborne particulate exposures in a home project type environment,” states Chambers.
Before you throw on an old N95 mask from your stash and start stripping the paint off your old furniture, you need to know what an N95 mask can’t protect you from.
“It is very important to recognize that N95 masks are not intended to offer protection against every type of atmospheric hazard,” states Chambers.
Oil particles: The “N” in N95 stands for “Not resistant to oil,” meaning that these masks will not filter out oil-based particles, such as those created by spraying pesticides or oil-based paint.
Ultra-small particulates: N95 masks cannot adequately filter particulates smaller than 0.3μm, such as metal or plastic fume.
Gases and vapors: N95s also do not provide any protection against gases, such as methane or carbon monoxide, or vapors, such as chemical fumes from solvents or paint.
Low-oxygen environments: N95s do not offer any protection in oxygen-deficient environments such as confined spaces, as they do not provide any oxygen to the wearer other than that in the surrounding air.
High-particulate environments: N95 masks will only offer protection from airborne particulates up to a point—they are not meant to be used for long periods of time in occupational settings that have extremely high levels of dust and other particulate.
How to Use an N95 Mask
A respirator mask will only protect you if it fits properly and forms a tight seal on your face. Some N95 respirators are available in different sizes, so be sure to select the right size for your face. If you have facial hair, such as a beard or sideburns, your mask will not be able to create a seal, and you will inhale particulates through the gaps between the mask and your skin.
When wearing your mask, make sure the straps are tight and the metal nosepiece fits snugly around your nose, with no gaps around your nose or cheeks. Always keep your nose inside the mask.
Furthermore, Chambers states, “N95 masks are disposable, meaning you should dispose of them when they become hard to breathe through, or if they get dirty, wet, or worn out. And do not try to wash them out and hang them to dry. Once they have done their job, throw them away.”
Below, we’ll discuss 9 DIY tasks that put you at risk of inhaling particulates that can harm your health over time with repeated exposure. Keep yourself safe during these activities by wearing an N95 respirator.
Stone, brick, tile, and other masonry materials contain crystalline silica, a common mineral found in many construction materials. Cutting, chipping, drilling, and grinding these materials casts silica dust into the air. These tiny particles are known as respirable crystalline silica. When you inhale silica dust, its particles can travel deep into your lungs, causing nodules and scar tissue to develop over time and resulting in silicosis, a serious and sometimes fatal lung disease. “Silicosis is associated with an increased risk of mycobacterial infection, chronic necrotizing aspergillosis, lung cancer, rheumatic disorder, kidney disease, chronic airflow obstruction, and chronic bronchitis,” states Dr. Mandapakala. NIOSH recommends wearing a respirator with a rating of N95 or better when exposed to silica dust.
2. Mixing Cement
Cement, the main component of concrete, also contains crystalline silica, making it an inhalation hazard in its dry form. You are most likely to be exposed to cement dust when mixing it or when emptying a bag of dry cement. Short-term exposure to cement dust can cause nose, throat, and upper respiratory system inflammation and difficulty breathing. Long-term exposure can lead to silicosis and other serious respiratory conditions. OSHA stipulates that workers wear a P-, R-, or N95 mask when working with concrete and that they should eat and drink only in dust-free areas to avoid ingesting cement dust.
3. Gardening
Gardening is good for the body and soul, but that doesn’t mean it it’s risk-free. Soil, compost, and potting mix can contain harmful bacteria, fungi, and other microorganisms. One such biological hazard is the bacterium Legionella longbeachae, found in potting mix and compost. L. longbeachae can cause Legionnaires’ disease, a severe form of pneumonia. Researchers have noted that those at higher risk of developing Legionnaires’ disease include smokers and those with chronic lung conditions. Wearing an N95 mask when working with compost or potting mix, along with good handwashing, can help you avoid inhaling or ingesting harmful microorganisms.
Inhaling wood dust over time has been linked to several symptoms and health conditions, from sneezing and breathing problems to asthma and cancer. Occupational exposure to hardwood dust in particular has been shown to cause cancer of the nasal cavity and paranasal sinuses. Dr. Mandapakala cautions, “Inhaling dust from activities like wood processing or wood sanding…can lead one to get asthma, COPD, and lung inflammation leading to interstitial lung disease and fibrosis and an increased risk for lung cancer.” The National Cancer Institute recommends reducing wood dust exposure in worksites through an exhaust ventilation system and by having workers wear respirator masks.
5. Drywall Sanding
Drywall sanding is a messy job, and it can also be a hazardous one. Drywall sheets and joint compound contain elements such as talc, gypsum, and silica, which become aerosolized through sanding or when the sheets are broken or disturbed. Breathing in drywall dust over time can cause throat and airway irritation, coughing, and breathing problems. Older drywall compound may also contain asbestos, so be sure to wear an N95 mask both when sanding new drywall and removing old drywall.
Blanket batts and rolls—the most prevalent type of household insulation—are commonly made from fiberglass. As indicated by its name, fiberglass is a synthetic fiber made of tiny particles of glass. These particles can irritate the skin and damage the respiratory system when they get lodged in the upper airway or inhaled deep into the lungs. Inhalation of fiberglass particles can result in irritation of the nose and throat and aggravation of existing asthma or other respiratory conditions.
The Insulation Contractors Association of America (ICAA) recommends cutting blanket insulation with a sharp knife rather than tearing or ripping it. ICAA also stipulates that workers installing or removing blanket insulation should wear a filtering facepiece respirator of N95 or greater.
7. Cleaning Animal Droppings
Animal droppings are a biological hazard because they harbor an array of pathogens harmful to humans. For example, raccoon latrines can be a source of Baylisacaris procyonis, a type of roundworm harmful to humans. Bird droppings can house Cyptococcus neoformans, a fungus that can infect humans when inhaled. And people can get infected with histoplasmosis when they breathe Hisoplasma fungal spores from bird and bat droppings. People who have weakened immune systems are particularly susceptible to severe forms of these infections. When cleaning up animal droppings around the home, always wear PPE, including a respirator mask.
8. Burning Yard Waste
Some municipalities allow you to burn brush or yard waste on your property. If burning is your practice to dispose of yard waste, take smoke exposure seriously. Smoke may smell nice, but it contains a varied mixture of gases and fine particles that can cause health problems when inhaled. Smoke inhalation has been shown to aggravate lung and heart conditions and even premature death in those with chronic diseases. If your municipality allows brush or yard waste burning, be sure to do it safely, in an open, well-ventilated area, and if you have a chronic lung or heart condition, wear an N95 respirator for good measure.
Molds are naturally present all around us and play an essential role in the breakdown of plant and animal matter. However, mold exposure can be harmful. For some people, inhaling mold spores may cause nasal congestion, coughing or wheezing, while those with asthma or mold allergies may have more severe reactions.
“Molds can cause asthma, bronchitis, hypersensitivity pneumonitis or infectious pneumonia, which can even progress to being necrotizing lung disease,” states. Dr. Mandapakala. The Centers for Disease Control and Prevention recommends wearing at minimum an N95 respirator mask when removing moldy items or cleaning up damage after a flood.
Understand the Limitations of Respirator Masks
While respirator masks like N95s can provide you with reliable, time-limited protection from many types of harmful particulates, face masks and other forms of PPE do not provide complete protection from harm. The best way to prevent harm from airborne hazards is to eliminate them at their source or avoid contact with them altogether. But if that’s not possible, keep a stack of N95 respirators in your home and workshop handy to help keep you—and your lungs—safe.
Dear Doctors: I work in a big shop where we make custom furniture. My wife thinks it puts me at risk of COPD and insists I should use a mask. Is she right? I thought COPD was something that happens to smokers. Plus, wearing a mask isn’t very comfortable.
Dear Reader: COPD is short for chronic obstructive pulmonary disease. It’s an umbrella term for a group of diseases in which damage to the tissues of the lungs, along with inflammation, obstruct the airways and make breathing difficult.
Symptoms include shortness of breath, a persistent cough, difficulty taking a deep breath, wheezing, excess mucus and a feeling of tightness in the lungs and chest. Because the airways of someone with COPD are obstructed, they can’t get enough oxygen. This causes an oxygen deficit in tissues throughout the body, which results in weakness, fatigue and a loss of stamina.
The two most common conditions associated with COPD are chronic bronchitis and emphysema. In chronic bronchitis, the lining of the bronchial tubes, which carry air to and from the tiny air sacs of the lungs, become inflamed. This causes excess mucus production and a chronic cough. It also puts the person at increased risk of having repeated respiratory infections. In emphysema, those tiny air sacs, known as alveoli, become permanently damaged. This leads to the oxygen deficit and resulting fatigue and breathing difficulties that we discussed earlier.
Smoking is a primary cause of COPD; The condition develops in response to repeated and long-term exposure to irritating gases and fine particulates, both of which smoking delivers in abundance. however, COPD can be an industrial hazard as well. People who work in occupations as varied as construction, mining, agriculture, welding, brick laying, stonemasonry, textiles, painting, and hair and nail care can all be at risk. When workers in these professions are also smokers, their chance of developing COPD goes up.
Occupational health data show that your own work in a carpentry shop, which exposes you to an environment that is not kind to the lungs, does put you at risk of developing COPD. The act of cutting, carving and sanding wood creates fine, airborne particulates that can damage the lungs and impair their ability to function. So can the fumes and gases emitted by the paints, stains, shellacs and solvents typically used in making furniture.
Even in a well-ventilated shop, particulates and gases will remain in the air. Long-term exposure to these can irritate, inflame and even damage delicate lung tissues, which can eventually lead to COPD.
COPD is a progressive disease. That means it gets worse with the passage of time. Although there is no known cure, it can be managed with medications and changes to behavior. Fortunately, you can significantly lower your own risk of developing this condition with one easy step: Always wear a high-quality, well-fitted mask while at work. It may be a bit uncomfortable, but to protect your lungs, it’s a small price to pay.
Dear Doctors: I work in a big shop where we make custom furniture. My wife thinks it puts me at risk of COPD and insists I should use a mask. Is she right? I thought COPD was something that happens to smokers. Plus, wearing a mask isn't very comfortable.
Dear Reader: COPD is short for chronic obstructive pulmonary disease. It's an umbrella term for a group of diseases in which damage to the tissues of the lungs, along with inflammation, obstruct the airways and make it difficult to breathe.
Symptoms include shortness of breath, a persistent cough, difficulty taking a deep breath, wheezing, excess mucus and a feeling of tightness in the lungs and chest. Because the airways of someone with COPD are obstructed, they can't get enough oxygen. This causes an oxygen deficit in tissues throughout the body, which results in weakness, fatigue and a loss of stamina.
The two most common conditions associated with COPD are chronic bronchitis and emphysema. In chronic bronchitis, the lining of the bronchial tubes, which carry air to and from the tiny air sacs of the lungs, become inflamed. This causes excess mucus production and a chronic cough. It also puts the person at increased risk of having repeated respiratory infections. In emphysema, those tiny air sacs, known as alveoli, become permanently damaged. This leads to the oxygen deficit and resulting fatigue and breathing difficulties that we discussed earlier.
You're correct that smoking is a primary cause of COPD. The condition develops in response to repeated and long-term exposure to irritating gases and fine particulates, both of which smoking delivers in abundance. However, COPD can be an industrial hazard as well. People who work in occupations as varied as construction, mining, agriculture, welding, brick laying, stonemasonry, textiles, painting, and hair and nail care can all be at risk. When workers in these professions are also smokers, their chance of developing COPD goes up.
Occupational health data show that your own work in a carpentry shop, which exposes you to an environment that is not kind to the lungs, does put you at risk of developing COPD. The act of cutting, carving and sanding wood creates fine, airborne particulates that can damage the lungs and impair their ability to function. So can the fumes and gases emitted by the paints, stains, shellacs and solvents typically used in making furniture.
Even in a well-ventilated shop, particulates and gases will remain in the air. Long-term exposure to these can irritate, inflame and even damage delicate lung tissues, which can eventually lead to COPD.
COPD is a progressive disease. That means it gets worse with the passage of time. Although there is no known cure, it can be managed with medications and changes to behavior. Fortunately, you can significantly lower your own risk of developing this condition with one easy step: Always wear a high-quality, well-fitted mask while at work. It may be a bit uncomfortable, but to protect your lungs, it's a small price to pay.
(Send your questions to [email protected], or write: Ask the Doctors, c/o UCLA Health Sciences Media Relations, 10960 Wilshire Blvd., Suite 1955, Los Angeles, CA, 90024. Owing to the volume of mail, personal replies cannot be provided.)
Samaritan’s Pulmonary Rehabilitation programs are designed to help people who have been diagnosed with asthma, COPD (chronic bronchitis, emphysema), bronchiectasis, fibrotic or interstitial diseases of the lung. Our staff of pulmonary rehab experts work together to develop specialized programs for you that combine exercise and education. Patients can attend up to 36 one-hour sessions per year, depending on their needs.
Education is a critical component to the Pulmonary Rehabilitation program. Patients will learn about:
Managing your symptoms and decreasing your problems with breathing.
Improving your physical condition and other factors impacting lung health.
Nutrition and weight loss.
Different lung diseases and the anatomy and physiology of the lungs.
The global landscape of respiratory health is undergoing a significant transformation, with the Asthma and COPD Drugs Market emerging as a crucial player in combating chronic pulmonary conditions. Valued at $32,988.7 million in 2020, this market is projected to soar to $52,049.54 million by 2030, marking a notable CAGR of 4.64% from 2021 to 2030.
Asthma, a chronic inflammatory lung disorder, and Chronic Obstructive Pulmonary Disease (COPD), characterized by irreversible airflow limitation, collectively pose a substantial burden on global health. Asthma, often triggered by allergies and environmental factors, manifests as recurrent wheezing, breathlessness, and chest tightness. COPD, primarily linked to tobacco smoking and occupational hazards, presents as a complex interplay of emphysema and chronic bronchitis.
Diagnosis of these conditions relies on a combination of physical examination and specialized tests such as X-rays and sputum analysis. Treatment modalities encompass a range of medications including inhaled corticosteroids, bronchodilators, and combination therapies tailored to manage acute exacerbations and provide long-term symptom control.
The burgeoning prevalence of asthma and COPD, as evidenced by WHO estimates indicating 262 million asthma cases and 46,1000 associated deaths in 2019, propels the growth of this market. Similarly, data from the American Lung Association highlighting 99 million adults with chronic bronchitis and 2 million with emphysema in the US alone in 2018 underscore the pressing need for effective therapeutic interventions.
Significant strides in respiratory disease management and the introduction of innovative pharmaceuticals further buoy market expansion. For instance, AstraZeneca's recent EU approval for 'Trixeo Aerosphere' for COPD maintenance treatment exemplifies the industry's commitment to addressing unmet clinical needs.
However, the market's trajectory is not without challenges. The exorbitant costs associated with asthma treatment serve as a barrier to access, inhibiting market growth during the forecast period.
Segmentation of the Asthma and COPD Drugs Market based on disease, medication class, and region offers valuable insights into market dynamics. While the asthma segment currently dominates due to rising patient numbers, the COPD segment is poised for robust growth fueled by therapeutic advancements.
Among medication classes, combination drugs lead the pack owing to their efficacy and convenience, although inhaled corticosteroids are expected to witness substantial growth driven by therapeutic innovations.
Geographically, North America commands the lion's share of the market, attributed to the high prevalence of asthma, established manufacturing infrastructure, and pervasive tobacco smoking habits. Nonetheless, Asia-Pacific emerges as a hotspot for market expansion, driven by burgeoning healthcare infrastructure and a burgeoning population.
Stakeholders stand to gain manifold from a comprehensive analysis of the Asthma and COPD Drugs Market, leveraging insights to identify investment opportunities and navigate strategic business decisions. Additionally, a thorough examination of key players and their growth strategies illuminates the competitive landscape, aiding stakeholders in charting their course amidst evolving market dynamics.
The Asthma and COPD Drugs Market presents a promising avenue for stakeholders amidst a backdrop of escalating respiratory health challenges. With innovative therapies, expanding markets, and strategic insights, stakeholders are poised to drive meaningful impact in the global fight against asthma and COPD.
Allied Market Research (AMR) is a full-service market research and business-consulting wing of Allied Analytics LLP based in Portland, Oregon. Allied Market Research provides global enterprises as well as medium and small businesses with unmatched quality of "Market Research Reports" and "Business Intelligence Solutions." AMR has a targeted view to provide business insights and consulting to assist its clients to make strategic business decisions and achieve sustainable growth in their respective market domain.
The full CMI on the next page has more details. If you are worried about using this medicine,
speak to your doctor or pharmacist.
1. Why am I using Symbicort Rapihaler?
Symbicort Rapihaler contains two active ingredients in one inhaler: budesonide and
formoterol (eformoterol) fumarate dihydrate. Symbicort Rapihaler is used for treatment
of asthma in adults and adolescents (12 years and over) or Chronic Obstructive Pulmonary
Disease (COPD) in adults (18 years and over).
2. What should I know before I use Symbicort Rapihaler?
Do not use if you have ever had an allergic reaction to any medicine containing budesonide
or formoterol, or any of the ingredients listed at the end of the CMI. Talk to your doctor if you have any other medical conditions, take any other medicines,
or are pregnant or plan to become pregnant or are breastfeeding.
Symbicort Rapihaler should be inhaled into your lungs through the mouth.
Follow all directions given to you by your doctor or pharmacist.
5. What should I know while using Symbicort Rapihaler?
Things you should do
If you have an Asthma Action Plan agreed with your doctor, follow it closely at all
times.
Have your reliever medicine available at all times. As advised by your doctor, this
may be your Symbicort Rapihaler (50/3 or 100/3) or another reliever medicine.
Rinse your mouth out with water after taking your daily morning and/or evening dose
of Symbicort Rapihaler and spit this out.
Remind any doctor, dentist or pharmacist you visit that you are using Symbicort Rapihaler.
Things you should not do
Do not stop using this medicine suddenly without checking with your doctor
Driving or using machines
Symbicort Rapihaler may cause dizziness, light-headedness, tiredness or drowsiness
in some people when they first start using it.
Looking after your medicine
Keep your Symbicort Rapihaler in a cool dry place where the temperature stays below
30oC, with the cover firmly in place.
Dispose your Symbicort Rapihaler 3 months after removal from the foil pouch.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of
them are minor and temporary. These include sore, yellowish, raised patches in the
mouth (thrush), hoarse voice, unpleasant taste in your mouth, pounding heart, headache,
trembling or muscle cramps. However, some side effects may need medical attention.
These include severe allergic reactions such as difficulty breathing, swelling of
the face, lips or tongue, severe rash or pneumonia (lung infection), signs include
fever or chills, increased phlegm or a change in colour, increased cough or difficulties
breathing. Serious side effects are rare. For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Active ingredient(s): budesonide / formoterol (eformoterol) fumarate dihydrate
Full Consumer Medicine Information (CMI)
This leaflet provides important information about using Symbicort Rapihaler. You should also speak to your doctor or pharmacist if you would like further information
or if you have any concerns or questions about using Symbicort Rapihaler.
Where to find information in this leaflet:
1. Why am I using Symbicort Rapihaler?
Symbicort Rapihaler is a pressurised metered dose inhaler (pMDI) or puffer. It contains
two active ingredients in one inhaler: budesonide and formoterol (as formoterol fumarate
dihydrate, which was previously known as eformoterol fumarate dihydrate).
Budesonide belongs to a group of medicines called corticosteroids. Budesonide acts
directly on your airways to reduce inflammation.
Formoterol belongs to a group of medicines called beta-2-agonists. Formoterol opens
up the airways to help you breathe more easily.
The medicine inside Symbicort Rapihaler is inhaled into the lungs for the treatment
of asthma in adults and adolescents (12 years and over) or Chronic Obstructive Pulmonary
Disease (COPD) in adults (18 years and over).
Asthma
Asthma is a disease where the airways of the lungs become narrow and inflamed (swollen),
making it difficult to breathe. This may for example be due to exercise, or exposure
to allergens (e.g. an allergy to house dust mites, smoke or air pollution), or other
things that irritate your lungs.
The budesonide in Symbicort Rapihaler helps to improve your condition and to prevent
asthma attacks from occurring.
The formoterol in Symbicort Rapihaler helps you breathe more easily.
Some people can take Symbicort Rapihaler when they need it – they use Symbicort Rapihaler
as an anti-inflammatory reliever to treat their symptoms when their asthma gets worse
and to help prevent asthma attacks, or to help prevent symptoms from happening (eg
before exercise or exposure to other triggers such as allergens).
Some people need to take Symbicort Rapihaler every day – they use their Symbicort
Rapihaler as a daily maintenance preventer to help maintain control of their asthma
symptoms and help prevent asthma attacks.
Chronic Obstructive Pulmonary Disease (COPD)
COPD (which includes chronic bronchitis and emphysema) is a long-term lung disease.
There is often permanent narrowing and persistent inflammation of the airways. Symptoms
may include difficulty in breathing (breathlessness or wheezing), coughing and increased
sputum (phlegm).
Symbicort Rapihaler when used as prescribed will help to control your COPD symptoms
(ie breathing difficulties).
2. What should I know before I use Symbicort Rapihaler
Warnings
Do not use Symbicort Rapihaler if:
you are allergic to any medicine containing budesonide or formoterol, or any of the
ingredients listed at the end of this leaflet. Always check the ingredients to make
sure you can use this medicine.
Check with your doctor if you:
have any allergies to any other medicines or foods.
have, or have had, any of the following medical conditions, as it may not be safe
for you to take Symbicort Rapihaler:
thyroid problems
diabetes
heart problems
liver problems
tuberculosis (TB)
low levels of potassium in the blood.
currently have an infection. If you take Symbicort Rapihaler while you have an infection,
the medicine may hide some of the signs of an infection. This may make you think,
mistakenly, that you are better or that it is not serious.
have any other medical conditions.
have any questions about how you should be using your Symbicort Rapihaler.
During treatment, you may be at risk of developing certain side effects. It is important
you understand these risks and how to monitor for them. See additional information
under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Your doctor will discuss the possible risks and benefits of using Symbicort Rapihaler
during pregnancy and while breastfeeding.
Children
Do not give Symbicort Rapihaler to a child under 12 years, unless directed to by the
child’s doctor.
Symbicort Rapihaler is not recommended for use in children under 12 years.
Asthma Action Plan
If you have asthma, ask your doctor or pharmacist if you have any questions about
your Asthma Action Plan.
Your healthcare professional should give you a personal Asthma Action Plan to help
manage your asthma. This plan will include what medicines to take as a reliever when
you have symptoms or sudden attacks of asthma, medicines you can take to prevent symptoms
from occurring (eg prior to exercise or allergen exposure) and if you need to take
daily maintenance medicines to help control your asthma. It will also provide advice
on when to seek urgent medical attention such as when your asthma suddenly worsens
or worsens over a period of time.
It is important that you discuss with your doctor both your exposure to triggers and
how often your exercise, as these could impact how your doctor prescribes your Symbicort
Rapihaler.
3. What if I am taking other medicines?
Some medicines may interfere with Symbicort Rapihaler and affect how it works. These
include:
medicines used to treat heart problems or high blood pressure such as beta-blockers,
diuretics and antiarrhythmics (disopyramide, procainamide and quinidine)
medicines used to treat glaucoma such as beta-blockers
medicines used to treat depression or other mood/mental disorders such as tricyclic
antidepressants, monoamine oxidase inhibitors and phenothiazines
medicines used to treat hayfever, coughs, colds and runny nose such as antihistamines
medicines used to treat fungal infections (eg ketoconazole)
xanthine derivatives (eg theophylline) which are a class of medicines used to treat
asthma and COPD
medicines used to treat Addison’s disease (when there is inadequate production of
a natural steroid hormone by the adrenal gland) or another condition where there is
too much salt lose in the urine (eg fludrocortisone)
These medicines may be affected by Symbicort Rapihaler or may affect how well it works.
You may need different amounts of your medicine, or you may need to use different
medicines. Your doctor or pharmacist will advise you.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins
or supplements you are taking and if these affect Symbicort Rapihaler.
Your doctor and pharmacist have more information on medicines to be careful with or
avoid while using Symbicort Rapihaler.
4. How do I use Symbicort Rapihaler?
How to use your Rapihaler
Follow all directions given to you by your doctor or pharmacist carefully.
They may differ from the information contained in this leaflet.
Each pack of Symbicort Rapihaler contains an instruction for use leaflet that tells
you the correct way to use it. Please read this carefully.
If you are not sure how to use the Rapihaler, ask your doctor or pharmacist to show
you how.
How much to take
Asthma (Adults and children 12 years and over)
Your healthcare professional should give you a personal Asthma Action Plan to help
manage your asthma. This plan will include what medicines to take as a reliever when
you have symptoms or sudden attacks of asthma, medicines you take prevent symptoms
from occurring (eg prior to exercise or allergen exposure) and if you need to take
daily maintenance medicines to help control your asthma.
It is important that you discuss with your doctor both your exposure to triggers and
how often you exercise, as these could impact how your doctor prescribes your Symbicort
Rapihaler.
Your doctor may have prescribed Symbicort Rapihaler for you to use as:
an anti-inflammatory reliever medicine only,
both an anti-inflammatory reliever and daily maintenance preventer medicine or,
as a daily maintenance preventer only, where another medicine is use as a reliever.
If your asthma has been under control for some time, your doctor may tell you to take
less inhalations of Symbicort Rapihaler, prescribe you a lower strength of Symbicort
Rapihaler or recommended that you use Symbicort Rapihaler in a different way.
If you are using more inhalations of your reliever medicine or you are wheezing or
breathless more than usual tell your doctor as your asthma may be getting worse.
Ask your doctor if you have any questions about how you should be using your Symbicort
Rapihaler.
Anti-inflammatory reliever only (Symbicort Rapihaler 100/3)
For patients aged 12 years and over, Symbicort Rapihaler 100/3 can be used to treat
asthma symptoms when they happen and to help stop asthma symptoms from happening (eg
just before exercise or before you get exposed to other triggers).
If you get asthma symptoms, take 2 inhalations and wait a few minutes. If you do not
feel better, take 2 more inhalations.
Your doctor will tell you how many inhalations to take before exercising or exposure
to other triggers to help stop symptoms from happening.
Do not use more than 12 inhalations on a single occasion or more than 24 inhalations
in any day. If your symptoms continue to worsen over 3 days, despite using additional
inhalations, tell your doctor.
Have your Symbicort Rapihaler reliever with you at all times.
Anti-inflammatory reliever plus maintenance therapy (Symbicort Rapihaler 50/3 and
100/3)
For patients aged 12 years and over, Symbicort Rapihaler 50/3 and 100/3 can be used
to treat asthma symptoms when they happen. Symbicort Rapihaler 100/3 can also be used
to help stop asthma symptoms from happening (eg just before exercise or before you
get exposed to other triggers).
If you get asthma symptoms, take 2 inhalations of Symbicort Rapihaler 50/3 or 100/3
and wait a few minutes. If you do not feel better, take 2 more inhalations.
Your doctor will tell you how many inhalations of Symbicort Rapihaler 100/3 to take
before exercising or exposure to other triggers to help stop symptoms from happening.
Have your Symbicort Rapihaler 50/3 or 100/3 reliever with you at all times.
You also need to take your Symbicort Rapihaler (50/3 or 100/3) daily as your maintenance
preventer. The usual maintenance dose is 4 inhalations per day (given either as 2
inhalations in the morning and evening or as 4 inhalations in either the morning or
evening). Your doctor may prescribe a maintenance dose of Symbicort Rapihaler 100/3,
4 inhalations twice a day.
Do not use more than 12 inhalations on a single occasion or more than 24 inhalations
of Symbicort Rapihaler (as needed and daily dose) in any day. If your symptoms continue
to worsen over 3 days, despite using additional inhalations, tell your doctor.
NOTE: Symbicort Rapihaler 200/6 is not recommended to be used as anti-inflammatory
reliever medicine.
For patients aged 12 years and over, Symbicort Rapihaler 50/3, 100/3 and 200/6 can
be used as a daily fixed-dose maintenance preventer.
The usual dose of Symbicort Rapihaler 50/3 and 100/3 is 2 or 4 inhalations twice a
day. Do not take more than 8 inhalations a day.
The usual dose of Symbicort Rapihaler 200/6 is 2 inhalations twice a day. Do not take
more than 4 inhalations a day.
Symbicort Rapihaler 200/6 can also be given as a higher dose in patients aged 18 years
and over. The usual dose is 4 inhalations twice a day. Do not take more than 8 inhalations
per day.
Have your separate reliever with you at all times.
COPD (Adults)
The usual dose (also maximum recommended dose) is 2 inhalations of Symbicort Rapihaler
200/6 twice a day.
Your doctor should tell you the best way to manage your symptoms and any flare ups.
This may include additional medicines (such as reliever medicines) to use when you
have sudden attacks of breathlessness.
If you are using more inhalations of your reliever medicine or you are wheezing or
breathless more than usual tell your doctor.
If your COPD gets worse, your doctor may give you some additional medicines (such
as oral corticosteroids or antibiotics).
How long to use your Symbicort Rapihaler
If your doctor has told you to take Symbicort Rapihaler daily, it is important that
you use it every day even if you feel well.
Symbicort Rapihaler helps control your asthma or COPD but does not cure it.
Keep using it for as long as your doctor tells you to. Do not stop using it unless
your doctor tells you to.
If you forget to use Symbicort Rapihaler
If you miss a dose of Symbicort Rapihaler, take your dose as soon as you remember.
Do not use a double dose to make up for the dose that you missed.
This may increase the chance of you getting an unwanted side effect.
If you are using Symbicort Rapihaler as a reliever medicine, consult your doctor on
the correct use of the product.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to use your medicine, ask your pharmacist for some
hints.
If you use too much Symbicort Rapihaler
If you think that you have used too much Symbicort Rapihaler, you may need urgent
medical attention.
You should immediately:
phone the Poisons Information Centre (by calling 13 11 26), or
contact your doctor, or
go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
If you use too much Symbicort Rapihaler, you may feel sick or vomit, have a fast or
irregular heartbeat, a headache, tremble, feel shaky, agitated, anxious, tense, restless,
excited or be unable to sleep.
5. What should I know while using Symbicort Rapihaler?
Things you should do
If you have an Asthma Action Plan that you have agreed with your doctor, follow it
closely at all times.
Keep using Symbicort Rapihaler for as long as your doctor tells you to, even if you
are feeling well.
See your doctor regularly to make sure that your asthma or COPD is not getting worse.
Have your reliever medicine available at all times. As advised by your doctor, this may be your Symbicort Rapihaler (50/3 or 100/3) or
another reliever medicine.
If you become pregnant while using Symbicort Rapihaler, tell your doctor.
Rinse your mouth out with water after taking your daily morning and/or evening dose
of Symbicort Rapihaler and spit this out. If you don’t rinse your mouth, you are more likely to develop thrush in your mouth.
You do not have to rinse mouth if you have to take occasional doses of Symbicort Rapihaler
for relief of asthma symptoms (ie as an anti-inflammatory reliever).
Call your doctor straight away if you:
are taking Symbicort Rapihaler for COPD and you notice any signs of pneumonia (infection
of the lung). Signs include fever or chills, increased phlegm/sputum production or
change in colour, increased cough or increased breathing difficulties. Pneumonia is
a serious medical condition and will require urgent medical attention.
Remind any doctor, dentist or pharmacist you visit that you are using Symbicort Rapihaler.
Things you should not do
Do not stop using this medicine suddenly without checking with your doctor.
Do not take any other medicines for your asthma or COPD without checking with your
doctor.
Do not give Symbicort Rapihaler to anyone else, even if they have the same condition
as you.
Do not use Symbicort Rapihaler to treat any other complaints unless your doctor tells
you to.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Symbicort
Rapihaler affects you.
Symbicort Rapihaler may cause dizziness, light-headedness, tiredness or drowsiness
in some people when they first start using it.
Looking after your medicine
Follow the instructions in the carton on how to take care of your medicine properly.
Storage
Keep your Symbicort Rapihaler in a cool dry place where the temperature stays below
30oC.
Always replace the mouthpiece cover after using Symbicort Rapihaler.
Discard Symbicort Rapihaler within 3 months after removal from the foil pouch.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do
not store it:
in the bathroom or near a sink, or
in the car or on window sills.
Keep it where young children cannot reach it.
WARNING
The canister in Symbicort Rapihaler contains a pressurised liquid. Do not expose to
temperatures higher than 50oC. Do not pierce the canister. The canister should not be broken, punctured or burnt,
even when it seems empty.
Cleaning
The Rapihaler mouthpiece must be wiped with a clean dry cloth/tissue and must never
get wet.
Full instructions on the right way to use and clean Symbicort Rapihaler are inside
each pack.
Getting rid of any unwanted medicine
Since some medicine may remain inside your Symbicort Rapihaler you should always return
it to your pharmacist for disposal including:
when you have taken all your doses and the dose counter is on zero (‘0’ – see instructions
in the pack), or
3 months after removal from the foil pouch, or
it is damaged or past its expiry date, or
your doctor/pharmacist has told you to stop using it.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of
them are minor and temporary. However, some side effects may need medical attention.
Tell your doctor or pharmacist as soon as possible if you do not feel well while you
are using Symbicort Rapihaler.
If you get any side effects, do not stop using Symbicort Rapihaler without first talking
to your doctor or pharmacist.
See the information below and, if you need to, ask your doctor or pharmacist if you
have any further questions about side effects.
Less serious side effects
Less serious side effects
What to do
Mouth/throat-related:
sore, yellowish, raised patches in the mouth (thrush)
hoarse voice
irritation of the tongue and mouth
coughing
These are less likely to happen if you rinse your mouth out after every time you use
your usual morning and/or evening dose of Symbicort Rapihaler.
Speak to your doctor if you have any of these less serious side effects and they worry
you.
Heart-related:
fast or irregular heart rate or pounding heart
chest pain
Nervous system-related:
feeling anxious, nervous, restless or upset
headache
trembling or shakiness
feeling light-headed or dizzy
thirsty
unpleasant taste in your mouth
tiredness
Gut-related:
nausea (feeling sick)
diarrhoea
Skin-related:
skin rash
skin bruising
Others:
difficulty sleeping
muscle twitching or cramps
weight gain
Speak to your doctor if you have any of these less serious side effects and they worry
you.
mood changes
Speak to your doctor if you notice any of these.
You may need urgent medical attention.
Serious side effects
Serious side effects
What to do
Allergic Reaction:
difficulty breathing or worsening of your breathing problems
swelling of the face, lips, tongue or other parts of the body
severe rash
Pneumonia (lung infection):
signs include fever or chills, increased phlegm/sputum production or a change in colour,
increased cough or difficulties breathing
Call your doctor straight away, or go straight to the Emergency Department at your
nearest hospital if you notice any of these serious side effects. You may need urgent medical attention. Serious side effects are rare.
Potential eye problem:
Any issues with your eyes such as blurred vision or other problems with your eyesight.
Speak to your doctor if you notice any of these.
Your doctor may need to send you to an ophthalmologist (eye doctor) to check that
you don't have eye problems such as cataracts (clouding of the eye lens), glaucoma
(increased pressure in your eyeballs) or other rare eye conditions reported with corticosteroids
use.
Other side effects
Growth
Corticosteroids taken into the lungs for long periods (eg 12 months) may affect how
children/adolescents grow. In rare cases, some children/adolescents may be sensitive
to the growth effects of corticosteroids, so the doctor may monitor a child's/adolescent's
height.
Tell your doctor or pharmacist if you notice anything else that may be making you
feel unwell.
Other side effects not listed here may occur in some people.
Some of these side effects (for example, changes in blood sugars) can only be found
when your doctor does test from time to time to check your progress.
Reporting side effects
After you have received medical advice for any side effects you experience, you can
report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems.
By reporting side effects, you can help provide more information on the safety of
this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop
taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Symbicort Rapihaler contains
Active ingredients
(main ingredient)
budesonide
formoterol (eformoterol) fumarate dihydrate
Other ingredients
(inactive ingredients)
apaflurane (HFA-227)
macrogol 1000
povidone
Do not take this medicine if you are allergic to any of these ingredients.
Symbicort Rapihaler does not contain lactose, sucrose, gluten, tartrazine or any other
azo dyes.
What Symbicort Rapihaler looks like
Symbicort Rapihaler is a pressurised metered dose inhaler with a dose counter. The
inhaler is comprised of a pressurised aluminum canister with an attached dose counter,
a red plastic casing body with a white mouthpiece and attached grey mouthpiece cover.
Each inhaler is individually wrapped in a foil laminate pouch (containing a sachet
of drying agent).
Symbicort Rapihaler is available in the following presentations*:
50/3: Each pack contains 1 inhaler of 120 inhalations of the medicine. [AUST R 158898]
100/3: Each pack contains 1 inhaler of 120 inhalations of the medicine. [AUST R 158899]
200/6: Each pack contains 1 inhaler of 60 (sample) or 120 inhalations of the medicine.
[AUST R 115555]
*not all presentations might be available in Australia
Who distributes Symbicort Rapihaler
AstraZeneca Pty Ltd ABN 54 009 682 311 66 Talavera Road MACQUARIE PARK NSW 2113
Telephone:- 1800 805 342
This leaflet was prepared on 11 Aug 2022.
® Symbicort Rapihaler is a registered trade mark of the AstraZeneca group of companies.
1Institute of Public Health, Gansu University of Chinese Medicine, Lanzhou, Gansu, People’s Republic of China; 2Department of Respiratory Medicine, Affiliated Hospital of Gansu University of Chinese Medicine, Lanzhou, Gansu, People’s Republic of China; 3The State Key Laboratory of Respiratory Disease, The First Affiliated Hospital, Institute of Public Health, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China; 4Institute of Basic Medicine, Gansu University of Chinese Medicine, Lanzhou, Gansu, People’s Republic of China
Correspondence: Yunchao Wang, Institute of Basic Medicine, Gansu University of Chinese Medicine, Lanzhou, Gansu, 730000, People’s Republic of China, Tel +86 18609404912, Email [email protected] Xinhua Wang, Institute of Public Health, Gansu University of Chinese Medicine, Lanzhou, Gansu, 730000, People’s Republic of China, Tel +86 13893602359, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is a chronic respiratory ailment influenced by a blend of genetic and environmental factors. Inflammatory response and an imbalance in oxidative-antioxidant mechanisms constitute the primary pathogenesis of COPD. Glutathione S-transferase P1(GSTP1) plays a pivotal role as an antioxidant enzyme in regulating oxidative-antioxidant responses in the pulmonary system. The activation of the NOD-like receptor thermal protein domain (NLRP3) inflammatory vesicle can trigger an inflammatory response. Several investigations have implicated GSTP1 and NLRP3 in the progression of COPD; nonetheless, there remains debate regarding this mechanism. Methods: Employing a case-control study design, 312 individuals diagnosed with COPD and 314 healthy controls were recruited from Gansu Province to evaluate the correlation between GSTP1 (rs4147581C>G and rs1695A>G) and NLRP3 (rs3806265T>C and rs10754558G>C) polymorphisms and the susceptibility to COPD. Results: The presence of the GSTP1 rs4147581G allele substantially elevated the susceptibility to COPD (CGvs.CC:OR=3.11,95% CI=1.961– 4.935, P< 0.001;GGvs.CC:OR=2.065,95% CI=1.273– 3.350, P=0.003; CG+GGvs.CC:OR=2.594,95% CI=1.718– 3.916, P< 0.001). Similarly, the NLRP3rs3806265T allele significantly increased the susceptibility to COPD (TC:TT:OR=0.432,95% CI=0.296– 0.630; TC+CCvs.TT:OR=2.132,95% CI=1.479– 3.074, P< 0.001). However, no statistically significant association was discerned between the rs1695A>G and rs10754558G>C polymorphisms and COPD susceptibility (P> 0.05). Conclusion: In summary, this study ascertained that the GSTP1 rs4147581C>G polymorphism is associated with increased COPD susceptibility, with the G allele elevating the risk of COPD. Similarly, the NLRP3 rs3806265T>C polymorphism is linked to elevated COPD susceptibility, with the T allele heightening the risk of COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a heterogeneous lung condition characterized by persistent respiratory symptoms and airflow limitation due to airway and/or respiratory disease, accompanied by dyspnea, cough, and sputum production.1–6 The global prevalence of COPD is 10.3%, with the prevalence among Chinese adults aged 40 years and older reaching as high as 13.6%, making it the third leading cause of death in the country.
The causes of COPD are complex, and its development is strongly associated with both genetic and environmental factors.7–9 Familial aggregation of COPD indicates the significant role of genetic factors in its development. Studies have indicated that only 10% to 20% of smokers eventually develop COPD, while 25% of COPD patients have never smoked.10–13 This suggests that COPD results from the interplay between genetic and environmental factors.14 Although the pathogenesis of COPD remains unclear, inflammation, protease-antiprotease imbalance, and oxidative-antioxidant imbalance are believed to be the primary mechanisms. Recently, Confalonieri et al propose to also include the concept of “structural changes due to failed regeneration by the distal airways progenitor cells” into the new definition of COPD “structural changes due to failed regeneration by the distal airways progenitor cells”.15 In recent years, several genetic polymorphisms have been linked to COPD development, including polymorphisms in glutathione S-transferase and inflammation-related genes.16,17
Glutathione S-transferase P1 (GSTP1) is a vital antioxidant enzyme highly expressed in the lungs.18,19 The GSTP1 gene, located on chromosome 11q13, has multiple single-nucleotide mutation sites.20 Polymorphisms at these mutant loci may impact their antioxidant function, leading to an oxidative-antioxidant imbalance that could induce COPD. Currently, there is controversy regarding the association between GSTP1 polymorphisms and COPD. While Yan and Ishii found an association, Yim and Yang’s study did not support a correlation between the two.21–26
The early immune response in the lungs against harmful stimuli is primarily mediated by inflammasomes, intracellular polyprotein complexes that recognize signals of injury and pathogens. NOD-like receptor thermal protein domain-associated protein 3 (NLRP3) is a crucial member of the inflammasome responsible for recognizing these signals.27 Mutations in the NLRP3 gene locus may result in abnormal inflammasome activation, leading to an excessive inflammatory response and exacerbating lung tissue damage.28–31 Faner et al and Eltom et al discovered that the rs35829419 mutation at the NLRP3 locus in lung tissues of COPD patients affects interactions with other proteins, further intensifying the inflammatory response and the release of inflammatory mediators.32,33 NLRP3 is also genetically polymorphic and has been linked to the development of several inflammatory diseases. However, although there are fewer studies on NLRP3 polymorphisms and COPD.34,35
In summary, COPD poses a heavy disease burden worldwide due to its high prevalence and mortality. However, the causes of COPD are complex, and its pathogenesis and causes have not been fully elucidated. Currently, it is mainly believed that the development of COPD is related to genes and environmental factors, and gene polymorphisms are also believed to be involved in many aspects of the pathogenesis of COPD. However, there is controversy at home and abroad about the association of GSTP1 gene polymorphisms with the risk of COPD and there are fewer studies on the association of NLRP3 gene polymorphisms with the risk of COPD. COPD is also less studied. Therefore, we conducted a case-control study in Gansu Province, including 312 COPD patients and 314 healthy controls, and used molecular epidemiological methods and real-time fluorescence quantitative PCR to analyse the association between GSTP1 and NLRP3 gene polymorphisms and COPD, with a view to providing theoretical basis for the early prevention programme and treatment of COPD-susceptible population.
Methods
Study Population
Between January 2020 and November 2022, we conducted a case-control study at the Affiliated Hospital of Gansu University of Traditional Chinese Medicine involving 312 COPD patients and 314 healthy controls from both rural and urban natural population cohorts in Gansu, the cases were mainly included in stable COPD patients with stable or mild symptoms such as cough, sputum, and shortness of breath. Our aim was to evaluate the association of GSTP1 (rs4147581C>G and rs1695A>G) and NLRP3 (rs3806265T>C and rs10754558G>C) polymorphisms in relation to COPD susceptibility.
Diagnostic Criteria and Pulmonary Function Tests for COPD
In accordance with the COPD Global Initiative for 2023, COPD is considered diagnosed when there are risk factors associated with dyspnea, cough, sputum production, and smoking, and the ratio of Forced Expiratory Volume in 1 second (FEV1) to Forced Vital Capacity (FVC), measured after a half-hour inhalation of 400 µg of salbutamol, falls below 70%. Pulmonary function tests were conducted using an EasyOne spirometer (NDD Medizintechnik AG, Switzerland) following the manufacturer’s instructions.36
Single Nucleotide Polymorphism (SNP) Selection and Genotyping
We identified potential risk SNPs by consulting the dbSNP database (www.ncbi.nlm.nih.gov/SNP) with the following selection criteria: SNPs located within the upper and lower 2000 bp of GSTP1 and NLRP3, Minor Allele Frequencies (MAFs) exceeding 0.05 in the Chinese population, and low linkage disequilibrium (LD, r2<0.8). Finally, we selected rs4147581C>G, rs1695A>G, rs3806265T>C, and rs10754558G>C for further analysis.
DNA extraction from collected peripheral blood samples was carried out using Tiangen Biochemical’s Blood Genomic DNA Extraction System Kit (DP349). Genotyping was performed using the TaqMan-MGB probe method. To ensure the reliability of PCR reactions, a negative control was included on each plate, and 10% of the samples were randomly selected for repeat testing.
Fluorescence-Based Quantitative PCR Technique
Mix configuration: Prepare PCR reaction mixture with Takara Premix Ex Taq™ (Probe qPCR) kit and vortex oscillation for 15 sec to mix the mixture well. Add sample: Standard DNA sample to 30 ng /μ l, add 18.5 μL PCR reaction mixture to 384 / 96 well plate, add l.5 μ l DNA template to 384 / 96 well plate, seal with sealing plate film and place at low speed, he centrifuwas centrifuged at 2000rpm for 2min. PCR amplification reaction: Place 384 / 96 well plate into 7900 HT quantitative PCR instrument / StepOnePlus quantitative PCR instrument and perform the following steps: Pre read → predenaturation 95°C 10min → denaturation 95°C 15s + annealing extension 60°C 1min (Repeat: 40 cycles) → 60°C 1min → Post read.
Methods of Statistical Analysis
Measurement data are presented as mean±standard deviation (X±SD), and count data are expressed as percentages (%). For group comparisons involving both categorical and continuous data, we employed χ2 tests and t-tests as appropriate.
In examining the association between genetic polymorphisms and COPD susceptibility, we utilized a multifactorial logistic regression model to calculate the odds ratio (OR) and its 95% confidence interval (CI). This analysis was adjusted for potential confounders such as age, gender, and smoking status. We conducted stratified analysis and interaction analysis for each stratum under each factor to assess the association between genotype and COPD risk. The Breslow-Day test was employed to evaluate heterogeneity between strata, with a significance level set at P<0.05. Statistical analysis was performed using SPSS version 25.0.
Results
Demographic Characteristics
Table 1 presents the demographic characteristics of our case-control study, including 312 COPD patients and 314 healthy controls in Gansu Province. Significant disparities in age, smoking status were observed between the two groups, with both demonstrating statistical significance (both P<0.05).
Table 1 Basic Demographic Characteristics of the Study Population
Association Analysis of SNPs in the GSTP1 and NLRP3 Genes with the Risk of COPD
As depicted in Table 2, the GSTP1 rs4147581 G allele markedly elevated the risk of COPD within the Gansu Province, China population (CGvs.CC:OR=3.11,95% CI=1.961–4.935, P<0.001;GGvs.CC:OR=2.065,95% CI=1.273–3.350, P=0.003;CG+GGvs.CC:OR=2.594,95% CI=1.718–3.916, P<0.001). In contrast, the GSTP1 rs1695A allele did not demonstrate a significant association with COPD risk. Nevertheless, logistic regression analysis, adjusting for age, gender, smoking, and BMI, revealed that the GSTP1 rs1695A>G allele was linked to an increased COPD risk within the Gansu Province, China population (AG:AA: adjusted Odds Ratio (OR)=2.495, 95% CI=1.558–3.996;AG+GGvs.AA: adjusted Odds Ratio (OR)=2.619, 95% CI=1.428–3.294, both P<0.001).
Table 2 Association of SNPs in GSTP1 and NLRP3 with COPD Susceptibility
The NLRP3 rs3806265T allele was also associated with COPD risk within the Gansu Province, China population (TC:TT: OR=0.432,95% CI=0.296–0.630; TC+CCvs.TT: OR=2.132, 95% CI=1.479–3.074, both P<0.001). Conversely, no significant association was detected between rs10754558G>C and the prevalence of COPD.
Stratified Analysis and Interaction Analysis of SNPs and Risk of COPD
Stratification and Interaction Analysis of GSTP1 rs4147581C>G and COPD Prevalence Risk
Table 3 presents the findings from the stratification and interaction analysis. Rs4147581C>G in the dominant model, when comparing the CC genotype to the GG/GC genotype, we observed variations in COPD risk across different strata defined by age, sex, and smoking history. Notably, there was no homogeneity between the strata (P<0.05). In the population aged over 60 years, the CC genotype (GG+GCvs.CC: OR=11.176,95% CI = 5.709–21.879) significantly increased the risk of COPD. Similarly, in the male population (GG+GCvs.CC: OR=22.784,95% CI=6.883–75.418), in individuals with a smoking history (GG+GCvs.CC: OR=24.429,95% CI=5.389–110.737), and in those without a smoking history (GG+GCvs.CC: OR=1.979,95% CI=1.327–2.950), the CC genotype was associated with an elevated risk of COPD. Furthermore, in individuals without a smoking history (GG+GCvs.CC: OR=1.327–2.950), the CC genotype exhibited an increased risk of COPD. Regarding BMI, marital status, alcohol consumption, and a history of previous diseases, we observed homogeneity between strata (P>0.05). In these strata, the CC genotype in the dominant model was associated with an increased risk of COPD: BMI (GG+GCvs.CC: OR=2.862,95% CI=1.968–4.162), marital status (GG+GCvs.CC: OR=2.950, 95% CI=2.031–4.286), alcohol consumption (GG+GCvs.CC: OR=3.046,95% CI=2.090–4.439), and a history of previous illnesses(GG+GCvs.CC: OR=2.898,95% CI=1.997–4.205).The results of multiplicative interaction analysis demonstrated that age and sex exhibited significant interactions with the genetic variation of rs4147581C>G, with a statistically significant difference observed between the case and control groups (P<0.05).
Table 3 Stratification Analysis and Interaction Analysis Between GSTP1 rs4147581C>G and COPD Susceptibility
Stratification and Interaction Analysis of NLRP3 rs3806265 T>C and COPD Prevalence Risk
The results of the stratification and interaction analysis are presented in Table 4. In the dominant model, rs3806265T>C, compared to the TT genotype, displayed variations in strata involving marital status and smoking. Notably, a lack of homogeneity between strata (P<0.05) was evident in the married population (TC+CCvs.TT: OR=2.816,95% CI=1.979–4.008), and the population with a smoking history (TC+CCvs.TT: OR=7.130,95% CI=3.291–15.451), and those without a smoking history (TC+CCvs.TT: OR=1.673,95% CI=1.156–2.421) increased the risk of COPD. In the dominant model, when stratified by TC/CC genotypes compared to TT genotypes, and across various strata, including age, sex, BMI, alcohol consumption, and history of previous diseases, we observed homogeneity between these strata (P>0.05). In each of these strata, the TC/CC genotypes were associated with an increased risk of COPD: age (TC+CCvs.TT:OR=2.088,95% CI=1.469–2.968), sex(TC+CCvs.TT:OR=2.268,95% CI=1.619–3.176), BMI(TC+CCvs.TT: OR=2.432,95% CI=1.743–3.392), alcohol consumption (TC+CC vs.TT: OR=2.339,95% CI=1.683–3.251), and history of previous illness (TC+CCvs.TT: OR=2.336,95% CI=1.680–3.247). Furthermore, the results of the multiplicative interaction analysis revealed that the interaction between smoking and alcohol consumption, in relation to the genetic variation in rs3806265 T>C, exhibited a statistically significant difference between the case and control groups (P<0.05).
Table 4 Stratified Analysis and Interaction Analysis Between NLRP3 rs380626 T>C and COPD Susceptibility
Analysis of the Relationship Between COPD Severity and SNPs
A total of 110 COPD patients with complete pulmonary function indicators and stable stage were selected from the case group. The severity of COPD was divided into four levels according to the percentage of the expected value of forced pulmonary capacity in the first second. Among them, the predicted value of FEV1 ≥ 80% was mild, and the predicted value of 50% ≤ FEV1 < 80% was moderate. 30% ≤FEV1 < 50% is expected to be severe, and FEV1 < 30% is expected to be extremely severe. The correlation between GSTP1 (rs4147581C>G and rs1695A>G) and NLRP3 (rs3806265T>C and rs10754558G>C) and COPD severity was analyzed. As shown in Table 5, the group of cases with complete lung function was analysed by chi-square test, and it was found that rs4147581C>G, rs1695A>G, rs3806265T>C, and rs10754558G>C were unassociated with the severity of COPD.
Table 5 Analysis of the Relationship Between COPD Severity and SNPs
Discussion
Gene polymorphisms are thought to be involved in several aspects of COPD pathogenesis, and the association between polymorphisms in the GSTP1 gene and the risk of COPD is controversial both nationally and internationally, and there have been fewer studies on polymorphisms in the NLRP3 gene and COPD. To elucidate the role of GSTP1 and NLRP3 polymorphisms in COPD development, our study examined the relationship between two SNPs in the GSTP1 gene (rs4147581C>G, rs1695A>G) and two SNPs in the NLRP3 gene (rs3806265T>C, rs10754558G>C) and COPD risk in the Gansu population, China. Notably, GSTP1 rs4147581C>G allele exhibited an elevated COPD risk, with CG and GG genotypes demonstrating higher susceptibility compared to the rs4147581CC genotype within the age>60 years, male, smoking population (P<0.05). Furthermore, the NLRP3 rs3806265T>C allele was linked to an increased COPD risk, with the TT genotype displaying a higher prevalence in the married and smoking populations (P<0.05) compared to the rs3806265TC/CC genotype. And neither rs4147581 nor rs3806265 was associated with COPD severity.
GSTP1, a member of the GST enzyme family, serves various physiological functions.37 GSTs have been reported to participate in antioxidant processes within the body.38 Among the GSTs, GSTP1 and its close association with oxidative stress and inflammatory responses in COPD are notable GSTP1 polymorphisms contribute to an imbalance in oxidative-oxidative processes, affecting the ability to counter oxidative stress and other lung disease-associated biological mechanisms. This, in turn, impacts COPD development and severity.39 Additionally, GSTP1 plays a protective role against airway cell damage induced by smoking. Consequently, the association between GSTP1 and COPD has garnered increased attention. NLRP3 protein primarily resides in cells, serving to detect in vivo danger signals, thus, promoting the release of inflammatory factors such as IL-1β and IL-18.40 NLRP3 plays a crucial role in macrophage and neutrophil migration and aggregation, as well as in oxidative stress generation.41 Inhibition of NLRP3 inflammatory vesicles indirectly mitigates the inflammatory effects of IL-1β and IL-18, presenting an ideal target for COPD treatment.
In this study, we selected two SNPs from the GSTP1 gene (rs4147581C>G and rs1695A>G) and two from the NLRP3 gene (rs3806265T>C and rs10754558G>C) for investigation. Online tools such as NCBI, Ensembl, and SNP info Web Server were used to obtain information on the location and function of these SNPs. The GSTP1 gene, located on chromosome 11q13, spans approximately 3 kb with 7 exons.42 Specifically, rs4147581C>G, positioned at Chr11: 67584114, has demonstrated significant associations with survival in patients with hepatocellular carcinoma, where its mutant genotype reduces the risk of death in these patients.43–46 Meanwhile, rs1695A>G at Chr11: 67585218 has shown associations with conditions such as cervical cancer, asthma, cyclophosphamide efficacy, and adverse effects. The NLRP3 gene, situated on chromosome 1q44, extends over approximately 40 kb.47,48 Within this gene, rs3806265T>C, located at Chr1: 247423034, has been linked to multiple sclerosis, juvenile systemic lupus erythematosus, and myasthenia gravis in recent studies.49–51 Similarly, rs10754558G>C at Chr1: 247448734 has been reported to be associated with rheumatoid arthritis, chronic kidney disease, psoriasis, and others.
Our study findings indicate an association between the GSTP1 rs4147581C>G variant and the risk of developing COPD. Patients with the CG/GG genotype exhibited a higher incidence of COPD compared to those with the CC genotype. GSTP1 functions as a detoxifying enzyme involved in intracellular oxidative stress responses and the removal of toxic metabolites.52 The rs4147581C>G variant may lead to reduced activity and function of the GSTP1 enzyme, diminishing its capacity to bind and eliminate toxicants and harmful metabolites. This can result in intracellular toxicant accumulation, increased oxidative stress, and exacerbated inflammatory responses, thereby elevating the risk of cardiovascular diseases like coronary artery disease, hypertension, and myocardial infarction.
Furthermore, we found an association between the NLRP3 rs3806265T>C variant and the risk of developing COPD, with a higher prevalence of the TT genotype compared to the TC/CC genotype.53 NLRP3 rs3806265 may play a pivotal role in inflammatory injury in COPD. The variant could lead to aberrant NLRP3 protein function, impacting the abnormal activation of inflammatory vesicles and deviating inflammatory signaling pathways. This, in turn, increases intracellular stress levels and contributes to the development and exacerbation of inflammatory diseases.
In this study, we investigated the role of GSTP1rs4147581C>G and NLRP3rs3806265T>C variants as risk factors for chronic obstructive pulmonary disease (COPD). Our findings provide a new way to predict and prevent COPD.54 In addition, studies by Yadav et al in North Indian populations found that GST gene polymorphisms can be used as susceptibility biomarkers for COPD, which also provides support for our study.55 The study of the effect of IL5RA variants on COPD in a Chinese population by Li et al provides new evidence to further our understanding of the genetic susceptibility to COPD. This study reveals an association between IL5RA gene variants and COPD, which could help us better predict and prevent COPD.56 The study of Castro et al on the influence of gene polymorphism on the severity of silicosis provides us with a new perspective. The study, conducted in silicon-exposed Brazilian workers, explored the effect of genetic polymorphisms on the severity of silicosis, providing useful information for the prevention and treatment of lung disease.57 Finally, Cheng et al ‘s study that circular RNA-SNPs may increase susceptibility to silicosis provides new clues to our understanding of genetic susceptibility to silicosis. The study, conducted in a Chinese population, identified a novel circular RNA-SNP that may increase susceptibility to lung lesions. Taken together, these studies provide valuable information for our understanding of the genetic susceptibility to COPD and lung-related diseases, which can help us better prevent and treat these diseases. In future studies, we will continue to explore the role of these gene variants in COPD, with a view to providing more guidance for clinical diagnosis and treatment.
This study explored the genetic role of GSTP1 and NLRP3 genes in the risk of developing COPD using a case-control design in Gansu Province. The results showed that genetic polymorphisms in these genes were correlated with the occurrence of COPD in Gansu Province. However, this study has its limitations. Firstly, the sample size is limited, and further validation with an expanded sample size is necessary. Secondly, the study subjects were sourced from specific hospitals, potentially introducing selection bias. Thirdly, there might be recall bias in the collection of past information during the questionnaire survey. Lastly, as this study is a case-control design, establishing the temporal sequence of genetic polymorphisms and COPD is challenging. Future research should consider cohort and experimental studies to further validate these findings.
Conclusion
In conclusion, GSTP1 rs4147581C>G allele exhibited an elevated COPD risk, with CG and GG genotypes demonstrating higher susceptibility compared to the rs4147581CC genotype within the age>60 years, male, smoking population. Furthermore, the NLRP3 rs3806265T>C allele was linked to an increased COPD risk, with the TT genotype displaying a higher prevalence in the married and smoking populations compared to the rs3806265TC/CC genotype.
Abbreviations
COPD, chronic obstructive pulmonary disease; GSTP1, Glutathione S-transferase P1; NLRP3, NOD-like receptor thermal protein domain associated protein 3; LD, Linkage disequilibrium; GWAS, genome-wide association analysis; SNPs, the single nucleotide polymorphisms; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; MAFs, the minor allele frequencies; PCR, TaqMan real-time polymerase chain reaction.
Acknowledgment
We thank the Institute of Public Health, Gansu University of Traditional Chinese Medicine. We thank the researchers in our laboratory for their guidance on experimental techniques.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the following grants: National Key Research and Development Project of China 2017YFC0907202 (Xinhua Wang); National Natural Science Foundation of China (82260889) (Xuhui Zhang); Central Guided Local Science and Technology Development Funds Project No.22ZY1QA003(Xinhua Wang); 2022 Gansu Province Higher Education Institutions Industry Support Program Project No.2022CYZC-53(Xinhua Wang).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Celli B, Fabbri L, Criner G, et al. Definition and nomenclature of chronic obstructive pulmonary disease: time for its revision. Am J Respir Crit Care Med. 2022;206(11):1317–1325. doi:10.1164/rccm.202204-0671PP
2. Soler N, Esperatti M, Ewig S, et al. Sputum purulence-guided antibiotic use in hospitalised patients with exacerbations of COPD. Europ resp J. 2012;40:1344–1353. doi:10.1183/09031936.00150211
3. Blakemore A, Dickens C, Chew-Graham CA, et al. Depression predicts emergency care use in people with chronic obstructive pulmonary disease: a large cohort study in primary care. Int J Chronic Obstr. 2019;14:1343–1353. doi:10.2147/COPD.S179109
4. Agustí A, Vogelmeier C, Faner R. COPD 2020: changes and challenges. Am J Physiol Lung Cell Mol Physiol. 2020;319:L879–L883. doi:10.1152/ajplung.00429.2020
5. Wang C, Xu J, Yang L, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): a national cross-sectional study. Lancet. 2018;391:1706–1717. doi:10.1016/S0140-6736(18)30841-9
6. Zhou M, Wang H, Zhu J, et al. Cause-specific mortality for 240 causes in China during 1990–2013: a systematic subnational analysis for the Global Burden of Disease Study 2013. Lancet. 2016;387:251–272. doi:10.1016/S0140-6736(15)00551-6
7. Kessler R, Partridge MR, Miravitlles M, et al. Symptom variability in patients with severe COPD: a pan-European cross-sectional study. Europ resp J. 2011;37:264–272. doi:10.1183/09031936.00051110
8. Montes de Oca M, Perez-Padilla R, Tálamo C, et al. Acute bronchodilator responsiveness in subjects with and without airflow obstruction in five Latin American cities: the PLATINO study. Pulmon Pharmacol Ther. 2010;23:29–35. doi:10.1016/j.pupt.2009.09.005
9. Silverman EK. Genetics of COPD. Annu Rev Physiol. 2020;82:413–431. doi:10.1146/annurev-physiol-021317-121224
10. He Y, Jiang B, Li LS, et al. Secondhand smoke exposure predicted COPD and other tobacco-related mortality in a 17-year cohort study in China. Chest. 2012;142(4):909–918. doi:10.1378/chest.11-2884
11. Forey BA, Thornton AJ, Lee PN. Systematic review with meta-analysis of the epidemiological evidence relating smoking to COPD, chronic bronchitis and emphysema. BMC Pulm Med. 2011;11:36. doi:10.1186/1471-2466-11-36
12. Eisner MD, Balmes J, Katz PP, et al. Lifetime environmental tobacco smoke exposure and the risk of chronic obstructive pulmonary disease. Environ Health. 2005;4:7. doi:10.1186/1476-069X-4-7
13. Wang B, Xiao D, Wang C. Smoking and chronic obstructive pulmonary disease in Chinese population: a meta-analysis. Clin Respir J. 2015;9:165–175. doi:10.1111/crj.12118
14. Lai HC, Lin TL, Chen TW, et al. Gut microbiota modulates COPD pathogenesis: role of anti-inflammatory Parabacteroides goldsteinii lipopolysaccharide. Gut. 2022;71:309–321. doi:10.1136/gutjnl-2020-322599
15. Confalonieri M, Braga L, Salton F, et al. Chronic obstructive pulmonary disease definition: is it time to incorporate the concept of failure of lung regeneration? Am J Respir Crit Care Med. 2023;207(3):366–367. doi:10.1164/rccm.202208-1508LE
16. Zhou H, Yang J, Li D, et al. Association of IREB2 and CHRNA3/5 polymorphisms with COPD and COPD-related phenotypes in a Chinese Han population. J Human Gene. 2012;57:738–746. doi:10.1038/jhg.2012.104
17. Yuan C, Chang D, Lu G, et al. Genetic polymorphism and chronic obstructive pulmonary disease. Int J Chronic Obstr. 2017;12:1385–1393. doi:10.2147/COPD.S134161
18. Stankovic M, Nikolic A, Nagorni-Obradovic L, et al. Gene-gene interactions between glutathione S-transferase M1 and matrix metalloproteinases 1, 9, and 12 in chronic obstructive pulmonary disease in serbians. COPD. 2017;14:581–589. doi:10.1080/15412555.2017.1369022
19. Zuntar I, Petlevski R, Dodig S, et al. GSTP1, GSTM1 and GSTT1 genetic polymorphisms and total serum GST concentration in stable male COPD. Acta Pharm. 2014;64:117–129. doi:10.2478/acph-2014-0003
20. Zhong L, Zhang YP, Fu WP, et al. The relationship between GSTP1 I105V polymorphism and COPD: a reappraisal. Am J Respir Crit Care Med. 2010;181:763–765. doi:10.1164/ajrccm.181.7.763
21. Yan F, Chen C, Jing J, et al. Association between polymorphism of glutathione S-transferase P1 and chronic obstructive pulmonary disease: a meta-analysis. Respir Med. 2010;104:473–480. doi:10.1016/j.rmed.2010.01.009
22. Ishii T, Matsuse T, Teramoto S, et al. Glutathione S-transferase P1 (GSTP1) polymorphism in patients with chronic obstructive pulmonary disease. Thorax. 1999;54:693–696. doi:10.1136/thx.54.8.693
23. Yim JJ, Park GY, Lee CT, et al. Genetic susceptibility to chronic obstructive pulmonary disease in Koreans: combined analysis of polymorphic genotypes for microsomal epoxide hydrolase and glutathione S-transferase M1 and T1. Thorax. 2000;55:121–125. doi:10.1136/thorax.55.2.121
24. Yang L, Li X, Tong X, et al. Association between glutathione S-transferase P1 Ile (105) Val gene polymorphism and chronic obstructive pulmonary disease: a meta-analysis based on seventeen case-control studies. Meta Gene. 2015;6:59–64. doi:10.1016/j.mgene.2015.08.007
25. Yang Q, Huang W, Yin D, et al. EPHX1 and GSTP1 polymorphisms are associated with COPD risk: a systematic review and meta-analysis. Front Genetics. 2023;14:1128985. doi:10.3389/fgene.2023.1128985
26. Du Y, Zhang H, Xu Y, et al. Association among genetic polymorphisms of GSTP1, HO-1, and SOD-3 and chronic obstructive pulmonary disease susceptibility. Int J Chronic Obstr. 2019;14:2081–2088. doi:10.2147/COPD.S213364
27. Mo R, Li J, Chen Y, et al. lncRNA GAS5 promotes pyroptosis in COPD by functioning as a ceRNA to regulate the miR-223-3p/NLRP3 axis. Molecul Med Rep. 2022;26:219. doi:10.3892/mmr.2022.12735
28. Faner R, Sobradillo P, Noguera A, et al. The inflammasome pathway in stable COPD and acute exacerbations. ERJ Open Res. 2016;2:2. doi:10.1183/23120541.00002-2016
29. Eltom S, Stevenson CS, Rastrick J, et al. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS One. 2011;6:e24097. doi:10.1371/journal.pone.0024097
30. Mahalanobish S, Dutta S, Saha S, et al. Melatonin induced suppression of ER stress and mitochondrial dysfunction inhibited NLRP3 inflammasome activation in COPD mice. Food and Chemical Toxicology. 2020;144:111588.
31. Zhang MY, Jiang YX, Yang YC, et al. Cigarette smoke extract induces pyroptosis in human bronchial epithelial cells through the ROS/NLRP3/caspase-1 pathway. Life Sci. 2021;269:119090. doi:10.1016/j.lfs.2021.119090
32. Cheng L, Yin R, Yang S, et al. Rs4612666 Polymorphism of the NLRP3 gene is associated with the occurrence of large artery atherosclerotic ischemic strokes and microembolic signals. Biomed Res. Int. 2018;2018:6345805. doi:10.1155/2018/6345805
33. Zhou D, Wang X, Chen T, et al. The NLRP3 rs10754558 polymorphism is associated with the occurrence and prognosis of coronary artery disease in the Chinese han population. Biomed Res. Int. 2016;2016:3185397. doi:10.1155/2016/3185397
34. Sui J, Li H, Fang Y, et al. NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in han Chinese. Arthritis Rheum. 2012;64:647–654. doi:10.1002/art.33370
35. Von Herrmann KM, Salas LA, Martinez EM, et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Parkinson’s Dis. 2018;4:24. doi:10.1038/s41531-018-0061-5
36. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2023 report). Avaliable from: goldcopd.org/2023-gold-report-2/. Accessed February7, 2024.
37. Gattás GJ, Kato M, Soares-Vieira JA, et al. Ethnicity and glutathione S-transferase (GSTM1/GSTT1) polymorphisms in a Brazilian population. Braz J Med Biol Res. 2004;37:451–458. doi:10.1590/s0100-879x2004000400002
38. Tomaki M, Sugiura H, Koarai A, et al. Decreased expression of antioxidant enzymes and increased expression of chemokines in COPD lung. Pulmon Pharmacol Ther. 2007;20:596–605. doi:10.1016/j.pupt.2006.06.006
39. Ishii T, Matsuse T, Igarashi H, et al. Tobacco smoke reduces viability in human lung fibroblasts: protective effect of glutathione S-transferase P1. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1189–L1195. doi:10.1152/ajplung.2001.280.6.L1189
40. Guo P, Li R, Piao TH, et al. Pathological mechanism and targeted drugs of COPD. Int J Chronic Obstr. 2022;17:1565–1575. doi:10.2147/COPD.S366126
41. Chen X, Liu G, Yuan Y, et al. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis. 2019;10:906. doi:10.1038/s41419-019-2157-1
42. Wang Z, Qu K, Niu W, et al. Glutathione S-transferase P1 gene rs4147581 polymorphism predicts overall survival of patients with hepatocellular carcinoma: evidence from an enlarged study. Tumour Biol. 2016;37:943–952. doi:10.1007/s13277-015-3871-7
43. Phuthong S, Settheetham-Ishida W, Natphopsuk S, et al. Genetic Polymorphism of the Glutathione S-transferase Pi 1 (GSTP1) and susceptibility to cervical cancer in human papilloma virus infected Northeastern Thai Women. Asian Pac J Cancer Prev. 2018;19:381–385. doi:10.22034/APJCP.2018.19.2.381
44. Mukhammadiyeva GF, Bakirov AB, Karimov DO, et al. Analysis of the GSTP1 rs1695 polymorphism association with the development of asthma and phenotypic manifestations. J Asthma. 2022;59:1065–1069. doi:10.1080/02770903.2021.1910295
45. Dasgupta RK, Adamson PJ, Davies FE, et al. Polymorphic variation in GSTP1 modulates outcome following therapy for multiple myeloma. Blood. 2003;102:2345–2350. doi:10.1182/blood-2003-02-0444
46. Zhong S, Huang M, Yang X, et al. Relationship of glutathione S-transferase genotypes with side-effects of pulsed cyclophosphamide therapy in patients with systemic lupus erythematosus. Br. J. Clin. Pharmacol. 2006;62:457–472. doi:10.1111/j.1365-2125.2006.02690.x
47. Imani D, Azimi A, Salehi Z, et al. Association of nod-like receptor protein-3 single nucleotide gene polymorphisms and expression with the susceptibility to relapsing-remitting multiple sclerosis. Int J Immunogene. 2018;45:329–336. doi:10.1111/iji.12401
48. Agah E, Nafissi S, Saleh F, et al. Investigating the possible association between NLRP3 gene polymorphisms and myasthenia gravis. Muscle and Nerve. 2021;63:730–736. doi:10.1002/mus.27193
49. Cheng L, Liang X, Qian L, et al. NLRP3 gene polymorphisms and expression in rheumatoid arthritis. Exp Ther Med. 2021;22:1110.
50. La Russa A, Lofaro D, Montesanto A, et al. Association between NLRP3 rs10754558 and CARD8 rs2043211 variants and susceptibility to chronic kidney disease. Int J Mol Sci. 2023;24:1110. doi:10.3892/etm.2021.10544
51. Dawood A, Shehata W. Evaluation of NLRP3 (rs10754558) and PTPN22 (1858C/T) (rs2476601) functional polymorphisms in psoriasis susceptibility in Egypt. Appl Clin Gene. 2021;14:331–339. doi:10.2147/TACG.S319065
52. Qu K, Liu SS, Wang ZX, et al. Polymorphisms of glutathione S-transferase genes and survival of resected hepatocellular carcinoma patients. World J Gastroenterol. 2015;21:4310–4322. doi:10.3748/wjg.v21.i14.4310
53. Kelley N, Jeltema D, Duan Y, et al. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20:3328. doi:10.3390/ijms20133328
54. Yadav H, Pandit D, Singh S, et al. GST polymorphism as a predictive biomarker for modulating the susceptibility to chronic obstructive pulmonary disease: a North Indian study. Exp Physiol. 2023. doi:10.1113/EP091339
55. Li S, Lin L, Zhao J, et al. The Study of the Influence of IL5RA variants on chronic obstructive pulmonary disease. COPD. 2023;20(1):338–347. doi:10.1080/15412555.2023.2270729
56. Castro MCS, Nani ASF, Salum KCR, et al. Genetic polymorphisms and their effects on the severity of silicosis in workers exposed to silica in Brazil. J Bras Pneumol. 2022;48(5):e20220167. doi:10.36416/1806-3756/e20220167
57. Cheng Z, Zhang Y, Zhao R, et al. A novel circRNA-SNP may increase susceptibility to silicosis. Ecotoxicol Environ Saf. 2022;242:113855. doi:10.1016/j.ecoenv.2022.113855
Breathing+ by Breathing Labs has passed peer review in a randomized controlled clinical trial that was recently published in SCI Q2 journal Pediatric Pulmonology. Research done by @bezmialem Full text is available in a link here: https://www.breathinglabs.com/clinical-trials/research-breathing-labs-and-nintendo-clinical-trial-is-published-in-journal-pediatric-pulmonology-sci-q2-impact-factor-3/?fbclid=IwAR2wNhSgurdbrrf3gzOOkHthgiWfXJ1x8RWvnMhkSo6fi33QPZEGzxzd6jM
BREAKING: @breathinglabs and @Nintendo clinical trial is published in journal Pediatric Pulmonology (SCI Q2, Impact Factor > 3), full text: https://breathinglabs.com/Nintendo%20&%20Breathing%20Labs%202022 #telemedicine #telehealth #mhealth
Clinical mouthpieces 10pcs packages are now available at 45€/50USD (shipping cost not included). Learn more: https://www.breathinglabs.com/latest-news/announcement-breathing-mouthpieces-for-clinical-and-professional-use-are-now-available/
BREATHING VR: Lately we are sourcing this VR headset for use in Breathing VR application. It allows easiest installation of both breathing+ headset cable, and USB charging cables, which is essential in professional use: https://www.banggood.com/VR-SHINECON-G5-VR-Glasses-3D-Virtual-Reality-Glasses-VR-Headset-For-iPhone-XS-11Pro-Mi10-p-1679808.html?rmmds=myorder&cur_warehouse=CN
Update: Each purchase of Breathing+ will now include three machine washable mouthpieces. Previous buyers will be supplied with those by their country representatives but will have to cover shipping costs. Please be patient while we arrange distribution. https://www.breathinglabs.com/latest-news/announcement-breathing-mouthpieces-for-clinical-and-professional-use-are-now-available/
Update: We moved servers + relocated all our games to our servers, please be patient while google reviews all that (showing unsafe website atm). Use duckduckgo or non-chromium browsers to reach our pages in the meantime. Everything ok + new product addons coming out in a month!
Registration and all functionalities at http://breathinglabs.com (and in our iOS and Android games) are fixed and fully working. If you find any issues -> [email protected]
We are back in stock with Breathing+, currently searching for VR supplier, and setting up mass production for toys and tens stimulation + in November we will be signing up new erasmus traineeships, research projects, bilateral, FP(eu), and asia-pacific ->[email protected]
BREAKING: Nintendo Co. Ltd (Japan) is implementing Breathing Games by @breathinglabs in FDA approved clinical trial for children with bronchiectasis: https://clinicaltrials.gov/ct2/show/NCT04038892
Notice to b2b partners: we are running late with some minor upgrade-> briefly running out of stock -> retail and b2b sale is closed until early october. To get a list of partners with stock to sell contact us at [email protected] Thanks, we'll go strong again in winter 💪