
Table of Contents
Study design
This is a pilot, exploratory, dose-adaptive, prospective, randomized, phase-2, open-label, proof-of-concept trial to assess the feasibility, safety, and efficacy of nebulized surfactant (Alveofact®, bovactant) in adult COVID-19 patients requiring mechanical ventilation (NCT04362059, date of registration 24/04/2020). The trial was conducted in the intensive care units at University Hospital Southampton (UK) and University College Hospitals London (UK)20. The study was sponsored by University Hospital Southampton (RHM CRI0399) and approved by the Health Research Authority (HRA), UK and Health Care Research Wales Ethics Committee (20/NE/0149), IRAS ID: 282498. Informed consent was obtained from all subjects and/or their legal representatives. The trial conducted in accordance with Good Clinical Practice (GCP), Health Research Authority (HRA) and Medicines and Health Care Regulatory Agency (MHRA, UK) regulations and standards. The reporting of this manuscript adheres to CONSORT guidelines. The study protocol is available as a supplementary file.
Eligible participants were 18 years of age or older, admitted to the intensive care unit and requiring invasive mechanical ventilation for acute respiratory failure following a positive respiratory sample real-time polymerase chain reaction (RT-PCR) SARS-CoV-2 test. All patients had radiological evidence of SARS-CoV-2 viral pneumonia and enrolled within 24 h of endotracheal intubation. The study was conducted between October 2020 and December 2021. Between 14th of October 2020 and 12th of November 2021, 77 patients were assessed for eligibility, and 20 patients were randomized. The University Hospital Southampton and University College Hospital London enrolled 13 and 7 participants respectively. One patient from the control group was transferred to the regional Extracorporeal Membrane oxygenation (ECMO) centre for additional support before study assessment at 48 h, leaving 19 participants for the primary analysis (Fig. 1).

Consort diagram for enrolment and randomization.
Trial procedures
Participants were randomly assigned to receive open-label nebulized surfactant or no intervention in a 3:2 ratio using an internet-based block randomization service (ALEA tool for clinical trials, FormsVision BV). As patients were unable to consent, consent was obtained from a personal legal representative (PerLR) or professional legal representative (ProfLR) prior to randomisation. Once regained capacity, all participants were approached for their consent for continued participation. The trial steering committee designed the protocol, and the oversight was provided by independent data and safety monitoring board (DSMB) and an external contract research organization (PHARMExcel). The trial protocol was previously published and registered in ClinicalTrials.gov (NCT04362059). All relevant data were collected and analyzed by the investigators. All the authors assume responsibility for the accuracy and completeness of trial data and the trial fidelity to the protocol and statistical analysis plan.
Surfactant composition
The recruiting centre did randomisation with a unique subject identifier specific to that centre. The nebulized surfactant is a natural lyophilized bovine surfactant (Alveofact®) preparation with an approximate composition of phospholipids [phosphatidylcholine (75%), phosphatidylglycerol (13%), phosphatidylethanolamine (3%), phosphatidylinositol (1%) and sphingomyelin (1%)], cholesterol (5%), surfactant proteins (1% SP-B and SP-C), and very low levels of free fatty acids, lysophosphatidylcholine, water, and 0.13% calcium. Each Alveofact® lyophilized vial consists of 108 mg of surfactant mixed at the bedside with a prefilled syringe containing 2.2 ml of 0.45% saline buffer for administration into the nebulizer.
Nebulizer device
This nebulizer device (Aerogen, Galway, Ireland) has a novel two-layer photo defined aperture plate (PDAP) vibrating mesh generating tiny surfactant droplets with mass median aerodynamic diameter < 3 µm to enhance distal lung deposition19. The nebulizer is controlled by two pole-mounted controllers with a sensor attached to the inspiratory limb of the ventilator to sense the start of the inspiratory phase and spray time adjusted to nebulize surfactant during the first 80% of the inspiratory phase of delivered breaths to optimize delivery and minimize wastage. The nebulizer is positioned between the ventilator circuit and the endotracheal tube (Fig. 2).
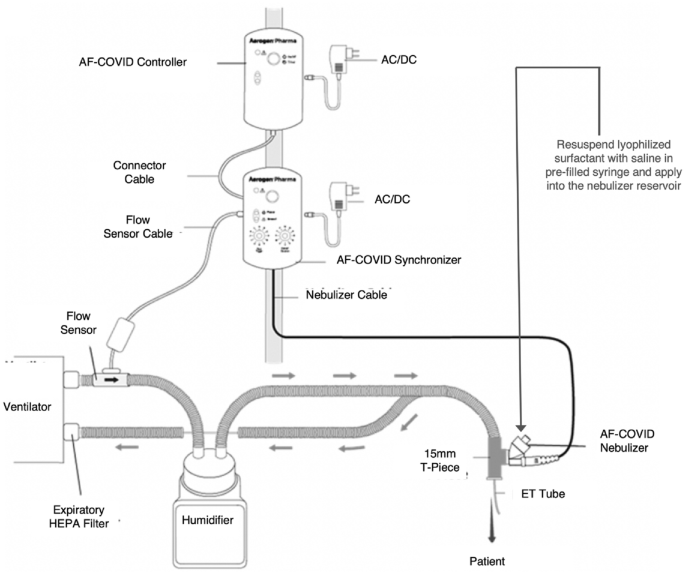
The nebulizer device and the ventilator circuit connections with the controller and breath synchronizer (modified from reference 19).
Nebulization dosing and timing
For the first 10-patient cohort, surfactant was administered at 0, 8, and 24-h post-randomization (Fig. 3). Due to the exploratory dose-finding nature of this study, the trial participants were divided into four cohorts with prospective graded dose escalation of total surfactant dose from 30 to 90 vials (3240–9720 mg). The first cohort received ten vials (1080 mg) at each dose scheduled at 0, 8 and 24 h. However, each vial took around 10–15 min to deliver, making it practically impossible to escalate the dose any further. The trial Data and Safety Monitoring Board (DSMB) was convened after this first cohort (N = 5) and advised the Trial Steering Committee (TSC) to proceed with the current dosing schedule for the second cohort, which was approved. Following completion of the second cohort (N = 5), biochemical analyses were conducted from the endotracheal aspirates, which confirmed an effective delivery but a rapid turnover with an estimated half-life for Alveofact phospholipid of ~ 7.6 h (range 1.8–20.8 h)11. Following further review by the DSMB and approval by the TSC, the dosing schedule was modified to provide a more frequent and prolonged surfactant administration with altered delivery timings as 0, 12, 24, 36, 48 h, and an optional dose at 72 h after randomization. In addition, the dose delivered was reduced from 1080 mg (10 vials) to 540 mg (5 vials). This dosing regime was maintained for cohorts 3 and 4 (Fig. 3).

Surfactant dosing regimen.
Exclusion criteria
The exclusion criteria were imminent expected death within 24 h, specific contraindications to surfactant administration (e.g. known allergy, pneumothorax, pulmonary haemorrhage), known or suspected pregnancy, stage 4 chronic kidney disease or requiring dialysis (i.e., estimated glomerular filtration rate < 30 ml/min), liver failure (Child–Pugh Class C), anticipated transfer to another hospital within 72 h (which is not a study site); current or recent (within 1 month) participation in another study that, in the opinion of the investigator, would prevent enrolment for safety reasons, and declined consent or assent. Patients were ineligible if they were intubated for more than 24 h.
Trial outcomes
The co-primary outcome was an improvement in oxygenation (PaO2/FiO2 ratio) and pulmonary ventilation defined as Ventilation Index (VI), where VI = [RR × PiP × PaCO2]/1000 at 48 h after study initiation. Ventilation index has been used widely by the paediatric surfactant replacement studies and provides a measure of the ventilation and incorporates variables from PaCO2, peak airway pressure and respiratory rate21,22,23. The secondary outcomes include frequency and severity of adverse events, change in pulmonary compliance, positive end-expiratory pressure (PEEP), ventilation index and PaO2/FiO2 ratio at 24 h after study initiation, clinical improvement defined by time to one improvement point on the ordinal scale described in the WHO master protocol (2020) recorded while hospitalised, duration of mechanical ventilation, mechanical ventilator-free days (VFD) at day 21, length of intensive care unit stay, number of days hospitalised and mortality at day 28.
Surfactant phosphatidylcholine assessment
Endotracheal aspirates through a closed in-line tracheal suction system were taken from all patients before surfactant nebulization and at 8, 16, 24, 48 and 72 h after recruitment. Samples were filtered and centrifuged at 400 g for 10 min at 4 °C. The supernatant was then lipid extracted by a modified Bligh and Dyer method24. Lipid extracts were analysed by electrospray ionisation mass spectrometry (ESI–MS/MS) (Waters Corporation, UK) to quantify surfactant phospholipids25,26.
Adverse event reporting
COVID-19 participants enrolled in this study were already critically ill with multiple COVID-19 related medical issues and were at risk of ongoing clinical deterioration and multi-organ failure. It was expected that many of these patients would experience events during their clinical pathway, but these were not reported unless the event was considered by the investigator to be associated with the study drug or delivery.
Any of the following pre-specified respiratory and cardiovascular deteriorations or adverse events occurring during nebulization and occurring within 48 h were recorded.
-
1.
Increase in oxygen (FiO2 ≥ 0.2 or more) or ventilator requirements (increase in PEEP of > 5 cmH2O or more to maintain target oxygenation).
-
2.
Sustained deterioration in pulmonary ventilation variables (> 10% increase in peak or mean airway pressures or decrease in tidal volume).
-
3.
Any episode of new cardiac arrhythmia
-
4.
Sustained increase in heart rate of > 20%
-
5.
Sustained reduction in mean arterial blood pressure (MAP) of > 10% or an increase in the vasopressor dose of Norepinephrine (0.1mcg/kg/min), epinephrine (0.1 mcg/kg/min) or the use of additional inotropes (dopamine/dobutamine/milrinone) or vasopressors (vasopressin/terlipressin/phenylephrine).
-
6.
New bronchospasm requiring treatment.
-
7.
Other respiratory deteriorations: pneumothorax (evidence on imaging), pulmonary haemorrhage (clinical) and acute lobar collapse (evidence on imaging), including transfer to tertiary hospital for ECMO were collected.
Statistical analysis
This is a pilot, exploratory dose-adoptive study and power calculations were based on significant dose response under varying assumed true dose response when using matched controls. Baseline data are presented as means (standard deviation) or medians (interquartile range) depending on the Normality of distribution. Categorical and binary variables are summarised as frequency and percentage of total. Baseline characteristics were compared between groups using independent samples t-tests for means, Mann–Whitney-U test for medians, and Fisher’s exact test for categorical data. Significance was defined at p < 0.05. For the co-primary endpoints of change in PaO2/FiO2 ratio and ventilation index, the difference between baseline and 48 h was calculated, and the Wilcoxon–Mann–Whitney test was used to test for the difference between groups. We then used quantile regression to adjust for the baseline value of the outcome variable. We also performed a secondary analysis of the primary endpoint splitting the surfactant group into cohorts 1&2 and cohorts 3&4, using the Kruskal Wallis test to investigate difference between the three groups (overall, cohorts 1&2, cohorts 3&4). The number of adverse events and their relationship to the study treatment were summarised as frequency and percentage of total, and risk ratios are reported with 95% confidence intervals. Surfactant phosphatidylcholine measurements are presented as percentage of total phospholipid as mean values and standard error of mean.

















