
[ad_1]
The year 2003 was an ominous one for emerging infectious diseases. A pair of deadly influenza strains had leapt from birds to humans in Hong Kong and the Netherlands. And a new coronavirus was spreading around the world causing a mysterious illness that became known as severe acute respiratory syndrome, or SARS. Many experts feared they were watching the start of a global pandemic.
Fortunately, the worst-case scenario never materialized. But it was a close-enough call for Robert Webster, a leading authority on avian influenza, to start urging scientists and policymakers to prepare for the next outbreak. One of his top recommendations: develop and stockpile drugs that target a wide range of viral pathogens1.
Drug researchers did not heed his call. After the SARS threat subsided, interest evaporated — and the world paid the price. “The scientific community really should have developed universal antivirals against SARS,” says Webster, now an emeritus member of St Jude Children’s Research Hospital in Memphis, Tennessee. “Then we would have had something in the stockpile for the emergence of COVID,” which is a caused by SARS-CoV-2, a close relative of the virus responsible for SARS.
Another warning shot came in 2012, when Middle East respiratory syndrome (MERS) — caused by another relative of SARS-CoV-2 — started spreading through a handful of countries. Still, the drug shelves remained largely bare — a fact that Jay Bradner, president of the Novartis Institutes for BioMedical Research in Cambridge, Massachusetts, regards as “regrettable”.
“Shame on us,” he says of the pharmaceutical industry. “We can be better prepared.”
Aside from one qualified success in remdesivir, a therapy originally developed to treat hepatitis C and Ebola, there were practically no strong antiviral drug candidates to quickly test and deploy against SARS-CoV-2. Researchers bemoan that there weren’t more options. “We need an arsenal,” says Kara Carter, head of discovery biology at the biotech company Dewpoint Therapeutics in Boston, Massachusetts, and president of the International Society for Antiviral Research.
New initiatives to create that arsenal are on the horizon. The US National Institutes of Health (NIH), for one, is planning a major programme to develop therapeutics against SARS-CoV-2 variants and other viruses with pandemic potential. A new industry-backed coalition is taking aim at influenza viruses and coronaviruses. And a few groups hope to create antivirals for more distantly related pathogens that pose a pandemic risk.
These projects won’t be starting from scratch. The past year has seen a bevy of SARS-CoV-2-centred drug-discovery efforts. But with the pharmaceutical industry historically focused on just a few particular viruses — HIV and hepatitis C, mostly — finding agents to fight known and imagined threats remains a tall order.
“There’s a lot of work that needs to be done,” says Nat Moorman, a virologist at the University of North Carolina (UNC) at Chapel Hill. But what choice does the scientific community have? “We don’t want to have another year like 2020,” Moorman says, “and we don’t have to, if we do the work in advance.”
Table of Contents
Ready for duty
Remdesivir came about thanks to the prudent forethought of researchers involved in the Antiviral Drug Discovery and Development Center (AD3C), an NIH-backed project launched seven years ago. Its objective is to screen existing drug libraries for inhibitors of influenza, coronaviruses, alphaviruses (such as those responsible for chikungunya), and flaviviruses (the pathogens behind dengue and Zika among others). In 2017, AD3C members demonstrated the anti-coronavirus potential of remdesivir in animal models2. Around the same time, trials that ran during two Ebola outbreaks in Africa showed that the drug was safe in people.

Laboratory technicians work on remdesivir at the Eva Pharma Facility in Cairo.Credit: Amr Abdallah Dalsh/Reuters
So, when COVID-19 hit, remdesivir was effectively ready to go. It could quickly go into human testing for the new coronavirus. In a large, placebo-controlled trial run over three months in early 2020, clinicians demonstrated that the drug accelerated recovery among people hospitalized with COVID-193. But remdesivir’s usefulness only goes so far. Some clinical studies have failed to confirm that it offers patients any benefit4. And the drug is expensive, difficult to manufacture and must be given intravenously in a hospital — all undesirable attributes in the middle of a pandemic.
Another antiviral drug now nearing approval could address some of those issues. Molnupiravir is an easier-to-synthesize, oral drug option that has been found to shorten the duration of infectiousness among people with symptomatic COVID-19. Late-stage clinical testing is under way.
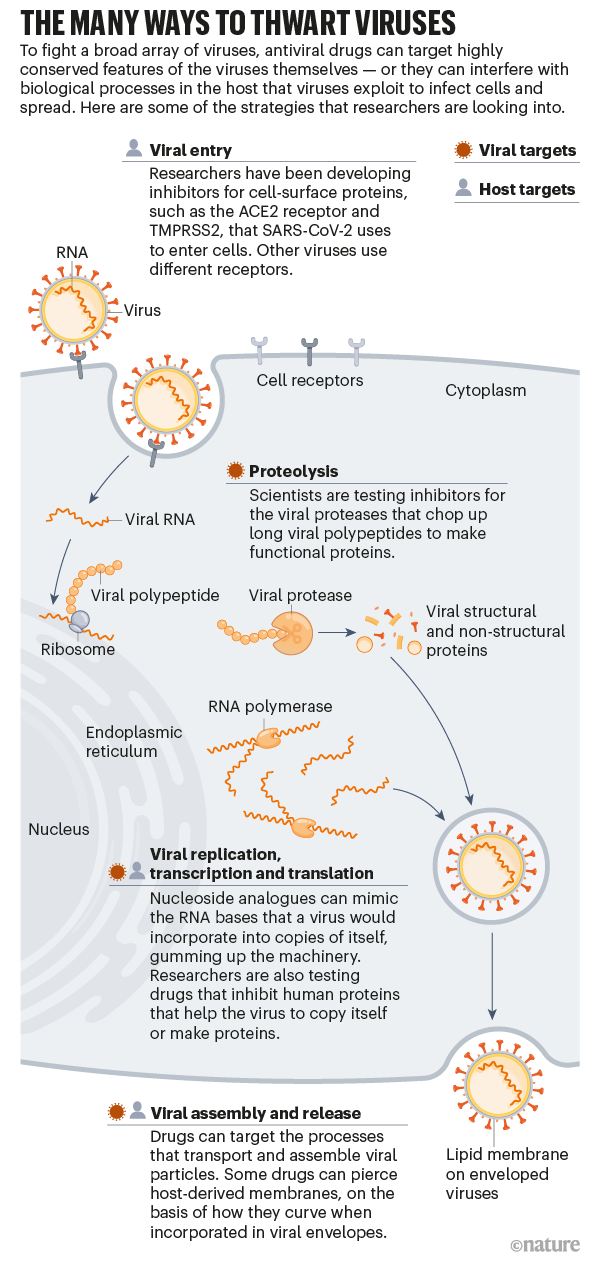
That drug, too, was first studied5 — pre-pandemic — by AD3C scientists, who have also identified promising leads against alphaviruses and flaviviruses. According to AD3C leader Richard Whitley, a paediatric infectious-disease specialist at the University of Alabama at Birmingham, all these drug candidates function as fake genetic building blocks that gum up the ability of viruses to copy their genomes faithfully. Instead of inserting the correct RNA bases during replication, a viral enzyme called polymerase is tricked into incorporating derivatives of the drugs. Human polymerases are unfooled, however, so only viruses are affected. (Similar drugs are used to treat hepatitis B, HIV and several other viruses.)
Because viruses in general are poor at catching genetic mistakes, these types of therapy — called nucleoside analogues — often work across viral families. Antiviral drugs that bind enzymes directly and block their function — which is to say, the vast majority of antivirals — do not typically have such broad activity. In principle, scientists could design drugs that work in many viruses by going after the most highly conserved regions of target proteins, says Jasper Fuk-Woo Chan, an emerging infectious-disease researcher at the University of Hong Kong. But, he adds, “traditionally, it’s always been a ‘one bug, one drug’ approach”.
That philosophy has served the industry well when it comes to making new medicines for HIV or hepatitis C. “But it’s proven inefficient in terms of rapidly addressing epidemics or pandemics,” Chan says.
Tricky targets
In many ways, the narrow activity of existing antivirals boils down to the nature of viruses themselves. Other types of pathogen — bacteria, fungi, parasites — are more easily contained because their cellular properties offer an abundance of targets for drug activity. Think about penicillin, which blocks cell-wall synthesis. Or azole antifungals, which disrupt the cell membrane.
Viruses, with their compact genomes and lack of cellular anatomy, offer many fewer druggable targets. Add in a high rate of replication — a typical SARS-CoV-2 infection, for instance, is thought to produce more than one million virions per person per day6 — coupled with an inherent genetic mutability, and it’s no wonder that most existing antivirals proved useless for COVID-19.
The plasticity of viruses means that a drug with activity against, say, herpes is unlikely to make a dent against a coronavirus. Alejandro Chavez, a bioengineer and antiviral drug researcher at Columbia University Irving Medical Center in New York City, thus doubts that anyone will find a “godly inhibitor that’s basically going to block everything”.
“What we will hopefully find”, he says, “are inhibitors that work on, if you’re really lucky, an entire family.” That would make the best-case scenario a pan-coronavirus inhibitor. But a more reasonable goal might be developing a drug for a subset of coronaviruses, such as alphacoronaviruses, which currently cause non-lethal infections in humans, and having a different drug for betacoronaviruses, the group responsible for SARS, MERS, and COVID-19.
Once the viral lineage is identified, “the same principles of drug discovery apply”, says Marnix Van Loock, head of emerging pathogens at Johnson & Johnson’s global public-health unit in Beerse, Belgium. As he explains, researchers need to find ‘druggable pockets’ on the surface of essential enzymes that are conserved between related viruses and can be used to design active molecules.
At least, that is, if the drug is directed at the virus itself. Some drug researchers instead aim to interfere with human pathways that a broad array of viruses commandeer for their own purposes. Jeffrey Glenn, for one, is developing a drug that blocks a fat-regulating enzyme used by many viruses to promote cellular entry and replication. By inhibiting this enzyme, “you deprive the virus access to a host function upon which it depends”, says Glenn, a gastroenterologist and molecular virologist at Stanford University School of Medicine in California.
Another host-directed antiviral strategy comes from two of Glenn’s former trainees — Nam-Joon Cho, a materials scientist at Nanyang Technological University in Singapore, and Joshua Jackman, a chemical engineer at Sungkyunkwan University in Seoul. They have developed small peptide drugs that poke holes in the lipid wrappings found around enveloped viruses7. These lipids come from the membrane surface of human cells. But the peptides penetrate only lipids that encase viruses, not cells, because of differences in the size of the membrane structure and how much it bends (see ‘The many ways to thwart viruses’).
Credit: Nik Spencer/Nature
Cho describes the lipid coating as the “common denominator” of all enveloped viruses — a group that includes flaviviruses, alphaviruses, coronaviruses, filoviruses, retroviruses and more. No other shared feature exists broadly across all those diverse viruses, which is why he thinks host-targeted antivirals might have greater potential as pandemic-preparedness tools.
Human biology also offers many more potentially druggable targets than do viruses. Plus, viruses are less able to develop resistance against host-targeted antivirals. A viral protein might need just a mutation or two to thwart drug binding, for example, whereas a host-targeted therapy could force the virus to exploit entirely different cellular processes.
Some scientists fear that tampering with human molecular pathways could cause unwanted side effects — but Shirit Einav, a virologist and infectious-disease specialist at Stanford University, thinks those toxicity concerns are overblown. “We treat every other disease by targeting a host function,” she says, and drug companies manage to find molecules and dosing regimens that people can tolerate. So, why should antivirals be any different? What’s more, she adds, “treating acute infections requires only several days of therapy” — not months or years, as for chronic illnesses — “which also helps reduce toxicity”.
Prep work
A combination of host-targeted and direct-acting drugs might offer the best insurance against future viral threats. But whatever strategy scientists pursue, experts agree that any drug intended for pandemic preparedness must, at a minimum, be fully tested in animal models and go through some trials in healthy human volunteers. “Then, when there’s a pandemic, we can rapidly deploy them at the best dose range in people,” says chemist Kelly Chibale, head of the Drug Discovery and Development Centre at the University of Cape Town, South Africa.
The goal would be to approve and distribute such a drug in the crucial window when other types of rapid-response medicine — such as vaccines or antibody treatments — are not yet available.
Drug developers took on some of that advance work in the wake of SARS. At drug company Pfizer’s La Jolla laboratories in California, for example, scientists responded to the 2003 outbreak by designing a molecule8 that inhibits a protein integral to coronavirus replication, an enzyme known as the main protease (Mpro), which chops up long chains of viral proteins into their functional parts.
For about six months, “it was a very intense effort”, says chemist Rob Kania, who led Pfizer’s SARS project. But infections soon petered out. And after the last cases of SARS were reported in 2004, Pfizer and other companies working on SARS drugs shelved their programmes. There just wasn’t a future market for the therapy. As UNC virologist Timothy Sheahan, who previously worked in pharmaceuticals, points out: “It’s hard to convince a company to make a drug against something that doesn’t exist.”
Kania’s team never had a chance to fully optimize its lead candidate for clinical use, let alone to test the therapy in mice or in people. So, when SARS-CoV-2 came along and genomic analyses revealed that the virus’s Mpro protein was almost identical to that from the original SARS pathogen, there was still a lot of chemical fine-tuning to do. By the time the drug, in a slightly different form, was ready for human testing9, the first wave of the pandemic had already subsided and almost one million people had died from the infection worldwide.
That drug, called PF-07304814, entered trials last September as an intravenously administered agent. Although the research could have been further advanced, at least Pfizer was not starting from scratch, says Charlotte Allerton, head of medicine design for the company. Although others are working to block the same target, Pfizer is the only drug maker with an experimental Mpro inhibitor in human testing today — two of them, in fact. Aside from its reformulated SARS drug, Pfizer started trials of a different oral candidate, PF-07321332, last month.
“Am I glad that we were in a position to move fast and that we’d done the pre-work? Yes,” says Allerton. “Do I wish we’d been even further down the line and been able to bring treatment options sooner? Absolutely.”
A wake-up call
Companies that hadn’t done the same kind of legwork are now pledging not to be caught empty-handed again. The pandemic has been “a wake-up call”, says John Young, global head of infectious diseases at pharmaceutical company Roche in Basel, Switzerland. “It’s just a matter of time before the next one,” he says, “and we need to prepare ourselves as an industry.”
To that end, leaders of the COVID R&D Alliance, a coalition of more than 20 life-sciences companies and venture-capital firms that came together last year to tackle SARS-CoV-2 collaboratively, are now launching a side project geared at broad-spectrum antivirals for coronaviruses and influenza viruses. According to Elliott Levy, the head of research and development strategy and operations at Amgen in Thousand Oaks, California, who is spearheading the effort, the group plans to advance around 25 candidate antivirals into initial human studies and build the clinical-trial infrastructure necessary for parallel testing when the next deadly virus strikes.
The US government has similar ambitions. Antivirals for coronaviruses are “task number one”, says NIH director Francis Collins. But, he told Nature, the initiative “certainly was intended to stretch to other viral families, if the funds are available”.
Complementary efforts come from the Corona Accelerated R&D in Europe project — a €75.8-million (US$90.1-million), five-year enterprise. It is geared at finding medicines both for the current COVID-19 pandemic and for future coronavirus outbreaks. Moorman and other UNC researchers, through their Rapidly Emerging Antiviral Drug Development Initiative, also hope to raise $500 million from governments, industry sponsors and foundations to develop broad-spectrum, direct-acting antivirals.
Meanwhile, some big pharmaceutical companies are ramping up their internal efforts. Novartis, for example, is now optimizing a pan-coronavirus inhibitor of the Mpro enzyme. According to John Tallarico, head of chemical biology and therapeutics at Novartis, the company is still at least a year away from human clinical testing, at which point COVID-19 might be well under control. Nonetheless, he says, Novartis is committed to moving this programme forward.
But, says Levy, “the level of investment from the industry today is not proportional to the threat” — which is why he hopes to raise around $1 billion from drug companies alone for the COVID R&D Alliance’s pandemic-preparedness spin-off venture. Extra funds could also come from non-profit organizations and other stakeholders, he says.
Andy Plump, president of research and development at Takeda Pharmaceutical in Cambridge, Massachusetts, and one of the leaders of the alliance, is optimistic about the programme’s chances of success. “Right now, you have a lot of energy behind this because there is the immediacy of SARS-CoV-2,” Plump says. But he doesn’t want apathy to set in again, like it did after SARS and MERS. “We need to lock in right now.”
[ad_2]
Source link

















